인코렌탈(incoRENTAL) 서비스로 CLC Genomics Workbench Premium의 Single Cell Module 일주일 간 활용해본 후기
안녕하세요, 이번 글에서는 인실리코젠(insilicogen)에서 운영하는 생물정보분석 단기 렌탈 서비스인 '인코렌탈(incoRENTAL)'에 대해 여러분께 소개해보고자 합니다.
- 본 글은 일정 수수료를 받고 일주일 간 인코렌탈 서비스를 체험하며 작성한 솔직 후기임을 밝힙니다 -

인코렌탈은 NGS 분석을 위해 여러 솔루션을 활용해야 할 때, 그러면서도 장기간 이용할 필요는 없어 단기간 동안만 이용하고자 할 때 유용한 서비스입니다.
인코렌탈 | (주)인실리코젠-생물정보 전문기업
요청하신 시험판 관련 문의는 정상 접수되었습니다. 연동된 계정으로 영업일 기준 2~3일 내 답변해드리도록 하겠습니다.
insilicogen.com
저는 인코렌탈이 제공해주는 솔루션 중 두번째 솔루션인 'CLC Genomics Workbench Premium(64 CPU / 256GB)'을 일주일 간 인코렌탈로 대여해 사용해보았습니다.

찾아보니까 'CLC Genomics Workbench Premium'은 퀴아젠(QIAGEN)이라는 다국적 독일 기업이 개발한 제품이더군요. 인실리코젠은 클라우드 서버를 구축하여 'CLC Genomics Workbench Preimum'과 같은 생물정보학 솔루션들을 사전에 설치해두고(+ 분석환경 구축) 원격 데스크톱을 통해 간편하게 본 솔루션들을 활용할 수 있는 인코렌탈 서비스를 운영하고 있습니다.
저는 단일세포 전사체분석에 관심이 있는 만큼 CLC Genomic Workbench Premium의 'Single Cell Module'을 사용해보았는데요. 활용 예시를 보여드리고자 간단하게 오픈 엑세스 데이터를 가져와 분석을 돌려봤습니다. 사용한 데이터는 작년에 '네이처 메디슨(Nature Medicine)' 저널에 퍼블리쉬된 논문에서 공개한 오픈 엑세스 scRNA-seq 데이터(FASTQ 파일 형태)입니다. 저는 그중에서 급성호흡곤란증후군(ARDS, acute respiratory distress)을 앓는 COVID-19 양성 환자 2명, COVID-19는 음성이지만 ARDS를 앓는 LRTD(하기도질환, lower respiratory tract disease) 환자 2명, LRTD와 COVID-19 모두 음성인 NO-LRTD 환자 1명 이렇게 총 5명의 blood 데이터를 활용했습니다. 데이터는 아래 링크의 사이트에서 가지고 왔습니다.
BioStudies < The European Bioinformatics Institute < EMBL-EBI
BioStudies – one package for all the data supporting a study
www.ebi.ac.uk
이미 진행된 연구를 재현해보는 것이고 샘플 수도 적은 만큼 새로운 의미있는 결과를 도출하기보다 활용 예시를 보여드리고자 합니다(본 분석에 있어 오류가 많을 수 있다는 점 양해 부탁드리겠습니다). 참고로 본 데이터를 오픈 엑세스로 공유한 연구1)에서는 말라리아 원충과 같은 기생충에 반복적으로 노출되는 사하라 이남 아프리카(SSA) 사람들은 코로나19(COVID-19) 감염 등의 질병에 있어 기생충 감염에 흔히 노출되지 않는 북반구 환자들과 다른 면역 균형을 보일 수 있다는 가설 하에 연구를 진행하였습니다. 그렇게 연구진은 말라위(Malawi) 성인 16명(코로나19 9명, 비‑코로나 LRTD 5명, 비‑호흡기 질환 2명)을 대상으로 폐, 간, 뇌, 혈액 등 여러 장기를 채취해 단일세포/단일핵 RNA‑시퀀싱(sc/snRNA‑seq) 등을 실시하였습니다. 앞서 말했듯 저는 그중 5명의 혈액 샘플로부터 얻은 scRNA-seq 데이터를 활용했습니다. 참고로 scRNA-seq과 같은 NGS 분석 기술을 이용하면 조건별로 등장하는 면역세포 유형과 세포별 유전자 발현 패턴 차이를 정량적으로 비교 분석할 수 있습니다.
[44일차] 단일세포 전사체 분석 기술 개요 및 프로세스 정리 01 :: sequencing library 준비 및 구축, illum
안녕하세요, 이번엔 단일세포 전사체 분석 기술(single cell RNA-seq analysis)의 전반적인 개요와 고려사항들을 정리해보는 시간을 가져보고자 합니다. 공부 겸 정리하는 건지라 오류가 있을 수 있다는
tkmstudy.tistory.com
이러한 개별세포 분석을 통해 우리는 특정 질환이 나타날 때, 혹은 질환의 중증도가 높아짐에 따라 어떤 세포의 어떤 기능에 교란이 나타나는지 확인하며 발병 메커니즘을 규명해나갈 수 있습니다. 물론 저는 몇 개의 샘플만 뽑아 데이터 분석을 해보는 것이고 현재 제 수준도 의미있는 결과를 낼 수준은 아닌 만큼 분석 프로세스 및 결과 제시에 있어 옳지 않은 부분이 있을 수 있으며, 그런 부분을 찾으신 분은 댓글 남겨주시거나 메일 보내주시면 교정하도록 하겠습니다.
Mail : tkm1214@naver.com
저는 앞선 논문에서 오픈 엑세스로 공개한 데이터를 FASTQ 파일(페어 엔드 형식) 형태로 다운 받았습니다. 본 데이터는 일루미나(illumina) 기기로부터 시퀀싱(Paired-end)되어 얻어진 '코딩 영역의 scRNA-seq 데이터'였습니다.

이제 인실리코젠에서 구축해준 CLC Genomics Workbench가 설치된 클라우드 서버에다가 다운받은 FASTQ 파일을 위치시켜보겠습니다. 이를 위해선 먼저 'FileZilla'라는 프로그램을 다운받아야 합니다. 프로그램을 실행하면 아래와 같은 화면이 나오는데요. 앞서 인코렌탈에서 제공해준 호스트 이름, 사용자명, 비밀번호, 포트(SFTP의 기본값은 22)를 입력하면 원격 연결이 되고, 파일을 옮겨 넣을 수 있습니다.

FileZilla를 활용해 다운받은 FASTQ 파일을 우분투 환경(인실리코젠이 구축한 서버)에 불러오는 방법은 아래 글에서 잘 설명해주고 있으니 참고 바랍니다.
인코렌탈 서비스를 이용한 NGS 데이터 분석 (feat. 사용방법, 장단점 및 후기)
오늘은 NGS 분석과 관련하여 인코렌탈이라는 생물정보 분석 솔루션 단기 임대 서비스에 대해 알아보겠습니다. 평소에 NGS 데이터를 통해 연구하거나 분석을 진행하는 분들께 유용한 서비스일 것
heestory533.tistory.com
이제 우분투 환경에 설치된 CLC Genomics Workbench를 실행하고, FileZila를 통해 우분투 환경에 위치시킨 데이터를 임포트(import)해보겠습니다. 임포트 방법은 데이터 형태마다 조금씩 다르지만, 일루미나 장비에서 얻은 페어엔드 시퀀싱 데이터(FASTQ 파일)는 홈 화면 좌측 상단에 있는 'import > illumina'를 선택하고 다음과 같이 두 리드의 FASTQ 파일을 'Add files'를 눌러 불러오면 됩니다.

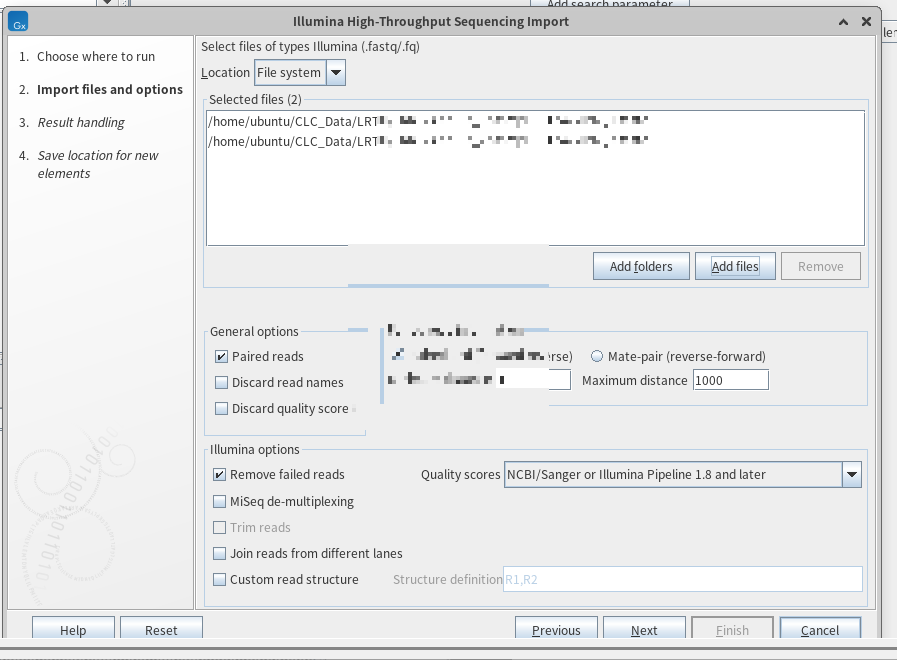
그러면 아래와 같이 품질 점수(Qualtiy Score)를 시각화해서 보여주는 '(paired)'로 끝나는 이름을 가진 게 등장할 겁니다. 그걸 클릭해주면 다음과 같은 화면이 나옵니다.

이제 불러온 FASTQ 파일에서 인서트(DNA 조각)만 남기고, 해당 리드에 '세포 바코드/UMI/Hashtag' 메타데이터를 부가하는 전처리를 진행해보겠습니다. 이를 위해 좌측 하단에 있는 Tool box에서 'Single cell Analysis', 그리고 거기서 'Annotate Single Cell Reads'를 클릭해줍니다.
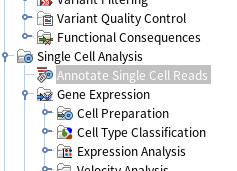
그럼 아래와 같은 화면이 등장하는데요. annotate할 싱글 셀 리드(임포트한 데이터)를 클릭해서 오른쪽으로 옮겨줍니다(만약 여러 개의 샘플을 사용할 경우엔 좌측 하단에 보이는 'Batch' 버튼을 눌러주어야 합니다).

그리고나서 Next 버튼을 누르다 보면 어떤 라이브러리를 사용했는지 선택하는 그런 화면이 등장합니다. 데이터 형태에 적합한 라이브러리를 선택하면 됩니다(ex. read1 : 바코드 및 UMI 서열, read2 : cDNA 서열). 그럼 좌측 맨 하단에 다음과 같은 글자가 뜨면서 annotate 절차가 진행됩니다.
프로세스가 끝나고 나면 각각의 데이터에 대한 'annotate된 결과 파일'과 '리포트 파일'이 생성이 되는데요. annotate가 잘 되었는지 확인하기 위해 리포트 파일 하나를 열어보겠습니다.

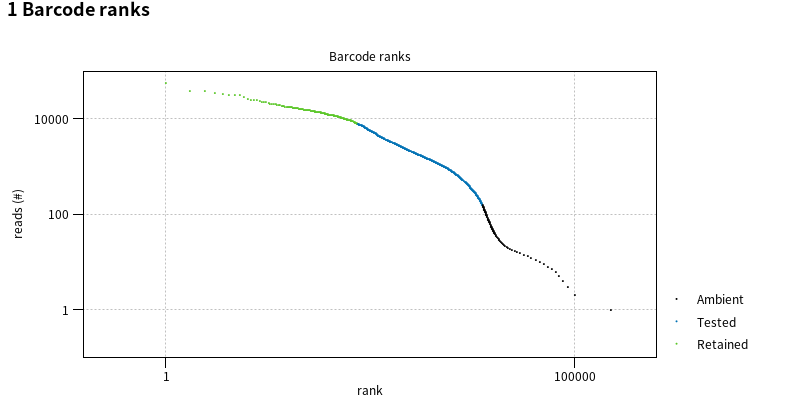
위의 표를 보면 바코드·UMI 구조가 규격과 맞지 않아 주석을 달지 못한 리드 수(Unmatched reads)는 없었음을 알 수 있습니다. 즉, 모든 리드에 대해 정상적으로 바코드 및 UMI 주석(annotate)을 완료했음을 알 수 있습니다. 또한, 일차적으로 매칭된 바코드 총 개수 중 시퀀싱 오류로 의심되어 '1-base mismatch' 혹은 'ambiguous(염기가 불확실한 'N'과 같은 상태)' 상태로 수정된 바코드 개수는 전체 중 약 4분의 1인 총 844,965개였다는 걸 알 수 있습니다. 최종적으로 오류 수정을 거쳐 유지된 바코드 수(Retained)는 수정된 바코드 개수를 뺀 3,028,694개였습니다. 리포트 밑으로 내려가면 'barcode ranks'가 등장합니다.
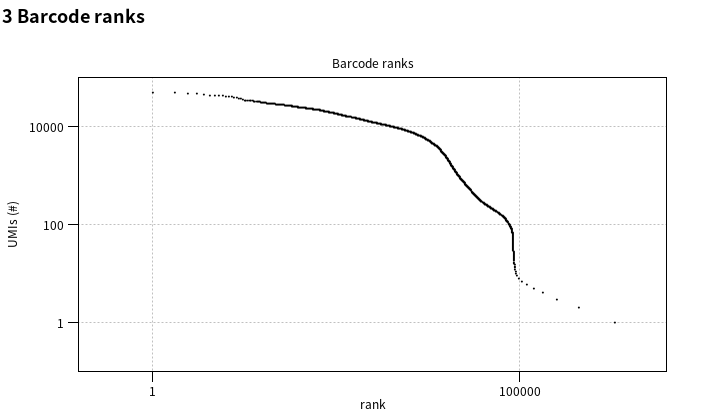
Barcode ranks는 세포 바코드를 총 UMI 개수에 따라 내림차순 정렬했을 때의 분포를 로그 스케일로 나타낸 것인데요. UMI 개수를 나타내는 y축을 기준으로 확인해보면, 1순위 주변의 세포(바코드)들은 수천~수만 개의 UMI를 보유한 반면, 오른쪽 끝 즉, 낮은 순위(10000위 주변)의 바코드들은 UMI 개수가 100개 이하까지 내려감을 확인할 수 있습니다.
하나의 바코드 내에서 UMI 개수가 100개 이하라는 건 하나의 세포 내에서 캡처된 RNA 개수가 100개 이하라는 의미입니다. 이는 보통 RNA들이 세포 내에서 캡처되었다기보다 세포가 없는 빈 드롭릿(empty droplet)에 '손상되었거나 죽은 세포에서 빠져나와 떠돌아다니는 ambient RNA들'이 100개 이하로 존재하는 것일 확률이 높습니다. 우리는 각 세포별로 어떤 유전자들이 얼마만큼 존재하는지 확인하고자 하기에 하나의 드롭릿에는 하나의 세포가 들어갔다는 전제 하에 연구를 진행해주어야 합니다. 따라서 뒤에 QC 단계에서 UMI 개수가 낮은 드롭릿들을 특정 기준에 따라 세포가 없는 빈드롭릿으로 간주하고 제거해줄 예정입니다. 다시 아래로 스크롤을 내리면 'Nucleotide counts for barcode positions' 즉, 셀 바코드로 지정된 리드 구간의 각 염기 위치별 A/T/C/G 빈도를 조사한 결과가 나옵니다.
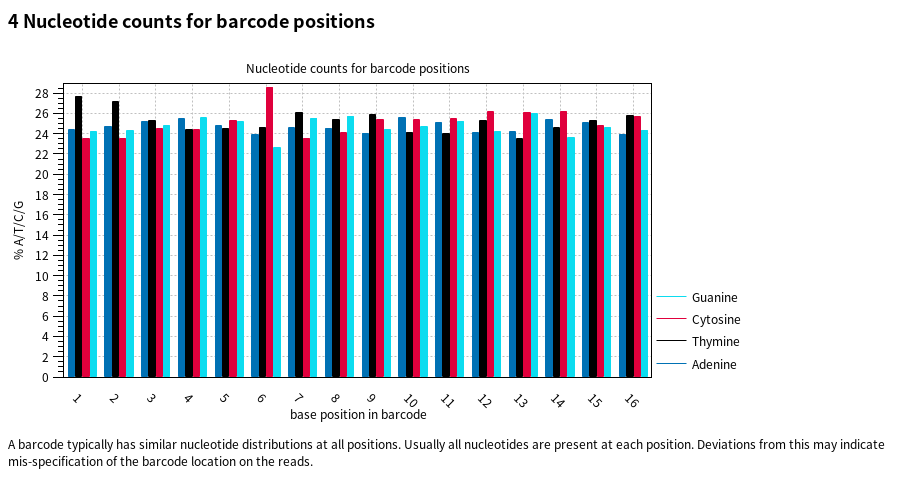
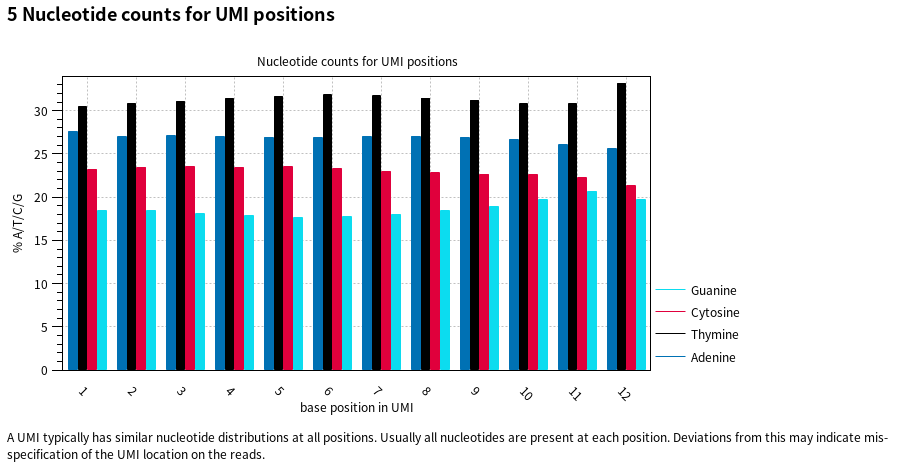
위의 영어 문장에 적혀있듯이 셀 바코드 라이브러리는 보통 각 위치마다 4개의 염기가 거의 동일한 비율로 섞여 있도록 설계되어 있습니다. 따라서 모든 위치에서 A/C/G/T가 약 20–30%씩 분포하는 것이 정상 신호라고 볼 수 있는데요. 위의 이미지를 보면 모든 위치에서 염기들이 24~28% 범위 내 분포해 있으니 셀 바코드 영역이 제대로 지정되었다고 볼 수 있겠습니다(만약 한 염기가 50% 이상 불균형하게 높았다면, 바코드 위치 설정을 재검토해야 할 수 있습니다). 더 아래를 내려가 보면 바코드 말고도 UMI 또한 위치별 뉴클레오티드 개수의 분포를 확인할 수 있으며 이 또한 균등하게 분포해야 UMI 영역이 올바르게 적용된 것으로 볼 수 있습니다. 아래 이미지를 확인해보면 모두 17~34% 범위이니 비교적 균일하게 분포함을 알 수 있습니다.
이렇게 annotate를 완료했으면 scRNA-seq analysis 단계로 들어갈 수 있는데요. 보다 정확한 결과를 얻기 위해서 그전에 트리밍 과정이 필요할 수 있습니다. 특히 페어 엔드 리드에서 시퀀서에 읽힌 어댑터(read-through adpaters)가 10% 이상 존재하면 '트리밍(Trimming)' 단계를 반드시 거쳐야 합니다3). 여기서 트리밍 단계는 낮은 퀄리티의 말단 서열이나 어댑터 서열을 제거해주는 단계입니다. 이러한 트리밍을 진행하려면, tool box의 'prepare sequencing data'에 있는 'Trim Reads'를 클릭하면 됩니다.

앞서 'annotate single cell Reads'에서 했던 방식으로 annotate된 두 개의 데이터를 오른쪽으로 옮겨줍니다(여러 샘플을 사용할 경우 좌측 하단의 Batch 버튼을 눌러주어야 합니다). 아후 Next를 누르면서 넘어가다보면, 트리밍 기준을 정할 수 있는 화면이 등장하는데요. 저는 디폴트 값인 Quality limit : 0.05와 Maximum number of amibiguites : 2로 설정했습니다. 이는 각각 말단 염기의 오류 확률이 0.05 이상(즉, Phred 품질 점수 기준 약 Q13 이하인 염기)인 경우 해당 염기를 제거하며, 불확실한 염기(N)가 2개를 초과하는 경우 해당 리드를 잘라내거나 제거한다는 의미입니다.
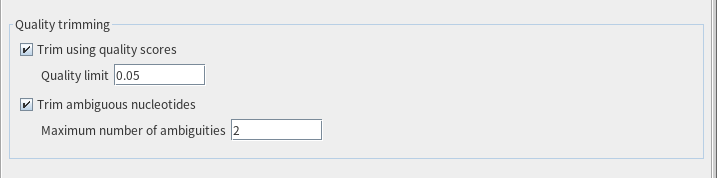
다음으로 저는 각 리드 쌍 양쪽 서열을 보고 시퀀서에 읽힌(read-through) 어댑터 시퀀스를 자동으로 파악하여 제거하도록 'Automatic read-through trimming'을 지정했습니다. 이렇게 설정해주고 나고 finishing 버튼을 누르면 트리밍이 진행되는데요. 만약 트리밍 후 말단에 남은 리드 길이가 분석 최소길이보다 짧다면 해당 리드는 폐기 됩니다. 트리밍 과정에 대한 자세한 설명은 IncoEDU 튜토리얼 1 강의 'Trim reads' 편2)에서 확인하실 수 있으니 참고 바랍니다. 이렇게 트리밍(Trimming)을 마치고 나면, 트리밍된 결과 파일들과 리포트 파일을 제공해줍니다.
간단하게 리포트 결과를 살펴보겠습니다. 먼저 '1. Trim Summary'를 보면 트리밍 전 리드 평균 길이는 90nt였는데, 트리밍 후에는 평균 길이가 5개 샘플 모두 89.7~89.8nt 쯤이 되었음을 알 수 있습니다. 즉, 평균 0.03~4 nt만 잘렸다는 걸 확인할 수 있었습니다.

또한, '4. Detailed trim results'에서 트림된 리드의 개수를 보니 상당수가 트리밍이 진행되긴 했지만, Ambiguity trim으로 제거된 534개의 리드들을 제외하고 다른 대다수의 리드들은 모두 유지됨을 알 수 있습니다. 마지막으로 'Automatic adapter read-through trimming'에 'No paired reads founds'가 적힌걸 보니 서열에 읽힌 어댑터 서열이 감지되지 않았음을 알 수 있습니다. 그만큼 시퀀서에 읽힌 어댑터 서열은 거의 없었다고 볼 수 있고, 추가적인 트리밍이 불필요하니 이제 해당 리드들을 가지고 본격적으로 scRNA-seq 분석을 진행해보도록 하겠습니다.

우선, 저는 리드 매핑 즉, alignment를 위해 필요한 본 플랫폼에서 제공해주는 사람 대상 참조 서열을 다운 받았습니다. 본 참조 서열 데이터는 우측 상단에 있는 'Manage Reference Data'를 클릭하고 분석하고자 하는 데이터에 적합한 참조 서열을 다운받으면 됩니다. 저는 single cell을 위한 human reference인 'Single Cell hg38(Ensembl)(GRch38)'을 다운받았습니다.


그럼 CLC_reference 디렉토리에 다음과 같은 파일들이 생성됩니다. 나중에 여기에 있는 참조 서열 데이터. 유전자 트랙, mRNA 트랙, 그리고 cell_type_classifier를 불러올 예정입니다.

이제 참조서열까지 다운받았으니 annotate되고, trimming된 리드 데이터들을 참조서열에 매핑해보겠습니다. 참고로 매핑(mapping)은 시퀀싱 데이터의 각 cDNA 서열들이 어떤 유전자의 서열에 해당하는지 이미 밝혀진 게놈 서열과 매칭해보면서 파악하는 과정으로, 이를 통해 각 세포에서 어떤 유전자가 얼마만큼 발현되어 나타나는지 정량적으로 확인할 수 있습니다. 매핑을 위해 다시 좌측 하단 tool box에서 'Single Cell Analysis > Gene Expression > Cell preparation > Single cell RNA-seq Analysis'를 클릭해줍니다.
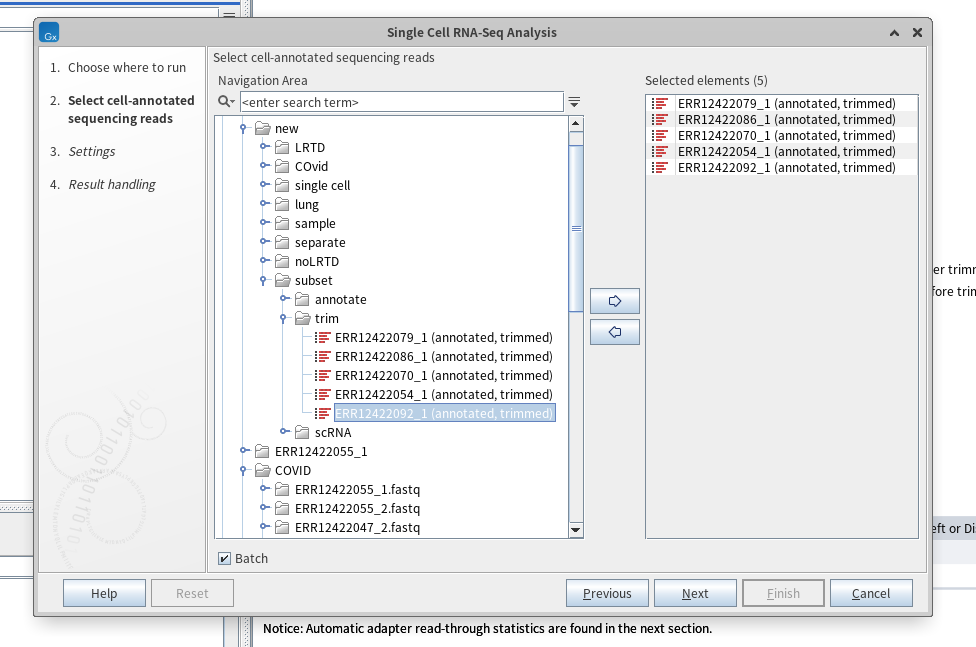
다음으로 앞서 다운 받은 'Single Cell hg38(Ensembl)' 파일의 각 데이터들을 reference sequence, Gene track, mRNA track 각각에 불러옵니다. 그리고 나서 다음 화면에서 'save'를 지정한 뒤 Finish 버튼을 누르면 매핑이 진행됩니다.

매핑 후에는 아래와 같이 '(RNA-seq report)'로 끝나는 파일과 '(GE)'로 끝나는 파일이 나옵니다. (GE)로 끝나는 파일이 매핑된 결과이고, report는 말 그대로 매핑 결과에 대한 보고서인데요. 그렇다면 report를 한번 열어 확인해봅시다.

여러 정보들이 나오지만, 그중에서 '4. Mapping statistics'를 확인해보면 전체 리드 중 몇 퍼센트가 매핑이 되었는지를 알 수 있습니다. 약 90%의 리드가 매핑이 된 걸 보니 매핑이 잘되었음을 알 수 있습니다. 만약 매핑이 50% 이하로 되었다면 오류가 났을 확률이 있습니다.

RNA-seq report 결과에 대한 자세한 해석은 아래 글을 참고하시길 바랍니다. 저는 글이 길어질 것 같으니 이것만 보고 넘어가겠습니다.
QIAGEN Bioinformatics Manuals
The Single Cell RNA-Seq Analysis report An example of an scRNA-Seq report is shown in figure 7.1. Figure 7.1: Report of an RNA-Seq run. The report is a collection of the sections described below, some sections included only based on the input provided whe
resources.qiagenbioinformatics.com
이제 매핑된 파일을 대상으로 QC(quality control)를 진행해보겠습니다. 'Cell preparation > QC for single cell'을 클릭하면 됩니다.

QC 절차에서는 빈드롭릿(empty-droplet), 더블릿(doublet), 그리고 품질이 낮은 세포가 들어간 드롭릿 등을 파악하여 필터링해주는데요. 각 단계가 어떤 단계이며 왜 필요한지는 아래 글에 소개해두었으니 참고바랍니다. QC 이후엔 정규화(Normalization), 특징선택, 차원축소, 클러스터링, 주석 순으로 데이터 분석이 진행되는데요. 이에 대한 설명도 아래 글에 해두었으니 참고바랍니다. 본 글에서는 해당 툴에서 각 단계들을 어떻게 실행하는지에 초점을 두어 설명하도록 하겠습니다.
[45일차] single cell RNA sequencing 기술 프로세스 정리 02 :: 시퀀싱 데이터 전처리 및 다운스트림 분석
안녕하세요, 이번 글에서는 저번 글에 이어 단일세포 전사체 분석 기술의 프로세스를 정리해보도록 하겠습니다. [44일차] 단일세포 전사체 분석 기술 개요 및 프로세스 정리 01 :: sequencing libra
tkmstudy.tistory.com
QC for single cell 툴을 클릭하면 다음과 같은 화면이 나옵니다. 앞서 만들어진 '(GE)'로 끝나는 파일을 오른쪽에 옮겨 넣습니다. 당연히 여러 샘플이니 Batch에 체크 표시를 해주어야 합니다.
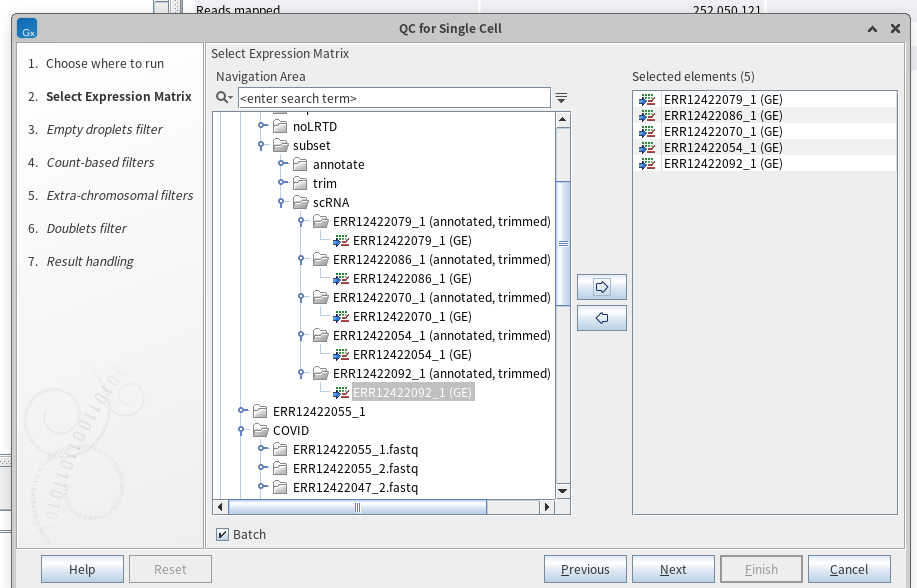
그러면 아래와 같은 화면이 나오는데요. 저는 빈 드롭릿(Empty Droplets)을 empty droplet filter로 필터링하도록 설정하였습니다. 또한, 비정상적으로 적은 혹은 비정상적으로 많은 리드 개수를 가진 드롭릿들을 필터링할 수 있는 기준(threshold) 또한 직접 계산해서 필터링하도록 지정해주었습니다.

전체 유전자 비율 중 미토콘드리아 유전자 비율이 상당 수준(ex. 5~10% 초과)으로 높다면, 그 세포는 세포자멸사(apoptosis)한 세포일 가능성이 높습니다. 세포자멸사를 하면 핵에 있는 유전자는 분해되지만 미토콘드리아의 유전자는 분해되지 않기 때문이죠. 따라서 보통 미토콘드리아 유전자 비율도 QC 기준으로 설정하며(보통 10%), 본 CLC GWB 플랫폼에서도 그러한 필터링이 가능합니다. 다음 화면으로 넘어가면 아래에서 처럼 'MT (mitochodria)'에 대한 feature tracks를 선택하라고 나오는데, 여기에 앞서 다운 받은 참조 데이터(hg38)의 Genes에 있는 파일을 불러와줍니다.

QC를 마치고 나면 다음과 같이 각각의 샘플에 대해 3개의 파일들이 등장합니다. QC report를 클릭해봅시다.
먼저 앞서 Trimmed report에 등장했던 'Barcode ranks' 분포가 등장하는데요. 리드 개수가 비정상적으로 적은 드롭릿들은 Ambient RNA만으로 구성된 빈 드롭릿으로 간주하고 필터링하였음을 알 수 있습니다.
이 뿐만이 아닙니다. ambient droplets를 어떤 기준으로 선택했는지도 리포트에서 상세히 알려주는데요. 예로 드롭릿에 세포가 들어있다고 간주 될 수 있는 최소의 리드 개수, 'ambient'로 간주할 수 있는 드롭릿이 가진 최대 리드 개수, 이를 통해 판별한 ambient droplet의 개수, 세포 당 리드 개수 중간 값 등 여러 요약통계량들을 표 형식으로 알려줍니다.

이외에도 쭉 아래로 내려가다보면 여러 QC 정보들을 제공해줍니다. 그중 Doublet(두 개의 세포가 하나의 드롭릿 안에 들어갔을 경우)은 얼마나 존재했는지 한번 확인해보겠습니다.

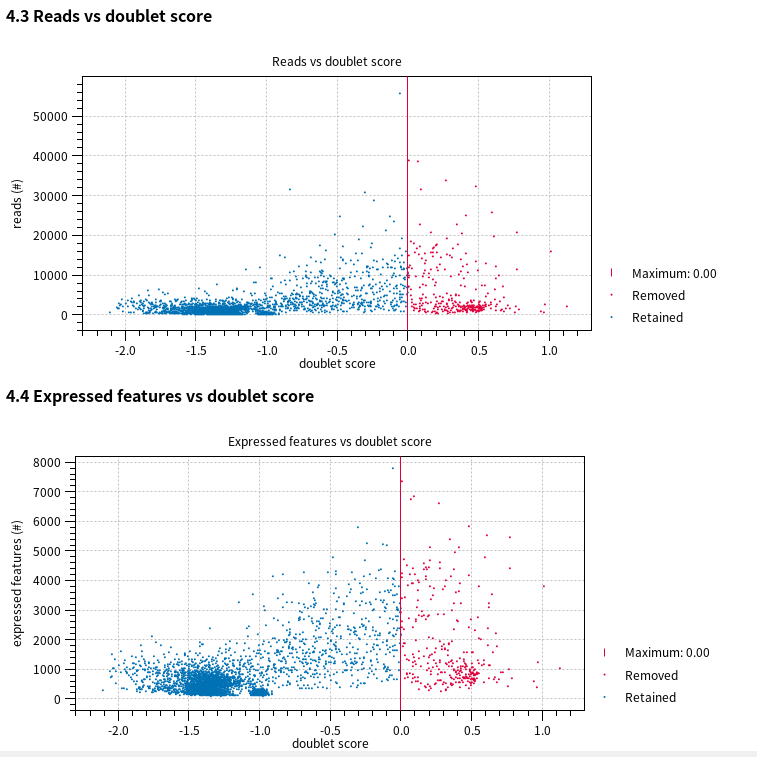
본 툴에서는 두 개의 드롭릿의 값(총 RNA 수, 유전자 개수)을 합친 artificial doublet을 만든 뒤, 각 드롭릿이 이 인공 더블릿과 얼마나 유사한지를 기준으로 doublet score를 매깁니다. 그리고 doublet score가 일정 기준 넘어가는 것들을 더블릿으로 간주하고 전체 5,602개의 세포 중 310의 세포를 제거했습니다(다른 샘플들도 확인하면 각각 더블릿으로 추정되는 드롭릿들을 제거했음을 알 수 있습니다). 그 외에도 다양한 QC 정보들을 제공해주는데 클릭 몇 번으로 이렇게 QC 결과들을 다양한 측면에서 깔끔하게 제공해주고, 필터링도 해주는 게 정말 편리합니다. 직접 코딩으로 하면 정말 번거로운 과정일 텐데 말이죠. QC 리포트 결과 해석은 아래 리포트를 참고하시길 바랍니다.
QIAGEN Bioinformatics Manuals
The output of QC for Single Cell The tool produces the following outputs: Note that for droplet-based protocols, each droplet is assigned one barcode and these terms can be used interchangeably. If the Empty droplets filter was enabled, the report contains
resources.qiagenbioinformatics.com
QC 이후에는 정규화(Normalization)를 진행해주어야 합니다. 정규화 단계는 한마디로 '세포 간 생물학적 차이의 비교'가 가능하도록 만들어주는 단계입니다. 정규화를 위해서는 'Normalize Single Cell Data' 툴을 사용합니다. 앞서 QC 결과로 얻은 '(filtered matrix)'로 끝나는 파일을 불러와줍니다. 이때는 Batch 버튼을 누르지 않아야 이후 5개의 샘플을 하나의 UMAP에 그릴 수 있는 것 같습니다.
결과적으로, 항상 그랬듯 툴로 처리(정규화)된 파일과 리포트 파일이 등장합니다. 리포트 파일을 보면, 정규화는 물론, 배치 보정, 그리고 HVG(Highly variable genes)까지 찾아냈음을 확인할 수 있습니다(HVG 이름에 앙상블 ID(ex. ENSG~)가 적혀있는건 구글 검색을 통해 어떤 유전자인지 알 수 있습니다).



정규화, 변동유전자 선정, 스케일링 및 회귀를 한번에 수행하는 글로벌 통계 모델링 접근법인 SCTransform이 떠오릅니다. 찾아보니 실제로 'Normalize Single Cell Data 툴이 SCTransform을 기반으로 알고리즘을 설계했다고 하네요. 더 궁금하신 분은 아래 글을 참고하시길 바랍니다. 빠른 진행을 위해 여기까지만 보고 넘어가겠습니다.
QIAGEN Bioinformatics Manuals
The Normalize Single Cell Data algorithm The algorithm is based on sctransform [Hafemeister and Satija, 2019]. Briefly, a negative binomial (NB) generalized linear model (GLM) is fit to genes, uniformly sampled across a range of expressions. The form of th
resources.qiagenbioinformatics.com
다음으로 SCTransform을 기반으로 정규화 절차가 완료된 데이터를 저차원 공간으로 축소해 UMAP 공간에 임배딩을 해보도록 하겠습니다(물론 국소적 구조에 강한 t-SNE도 사용할 수 있는 툴이 본 플랫폼 내에 존재합니다). UMAP(Uniform Manifold Approximation and Projection)은 비선형 차원 축소 알고리즘으로, 2차원 시각화를 진행할 수 있습니다. 즉, 세포 간 유전자 발현의 유사도를 기준으로 고차원에서 서로 가까운 거리에 있는 점(세포)들은 저차원에서도 가깝게, 멀리있는 점들은 멀게 배치하여 데이터의 내재적 구조를 보존하면서 저차원 공간으로 차원 축소를 진행하는 것이죠. 이러한 방식으로 UMAP plot을 그리기 위해 'dimensionality reduction > UMAP for single cell'을 클릭해줍니다. 그리고 나면 아래와 같은 화면이 나옵니다. 여기서 앞서 정규화한 '(residuals)'로 끝나는 파일을 오른쪽에 옮겨줍니다.
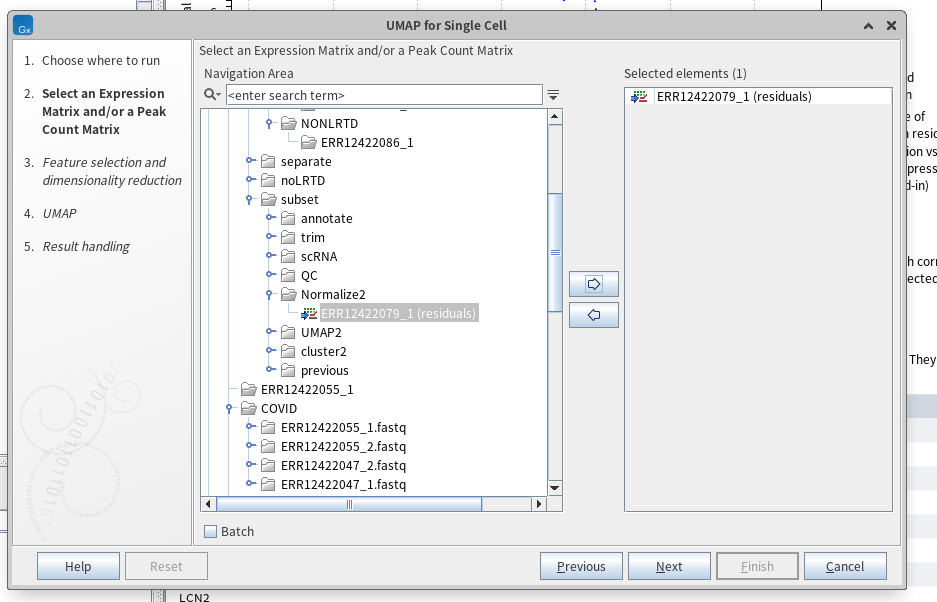
저는 HVG 3000개, 주성분 20 차원, 그리고 아래와 같은 threshold를 기준 값으로 설정하였습니다. UMAP을 여러 차례 그려보면서 설정한 기준 값인데, 연구자 재량껏 데이터의 특성과 연구 목적에 맞게 설정해주시면 될 것 같습니다.

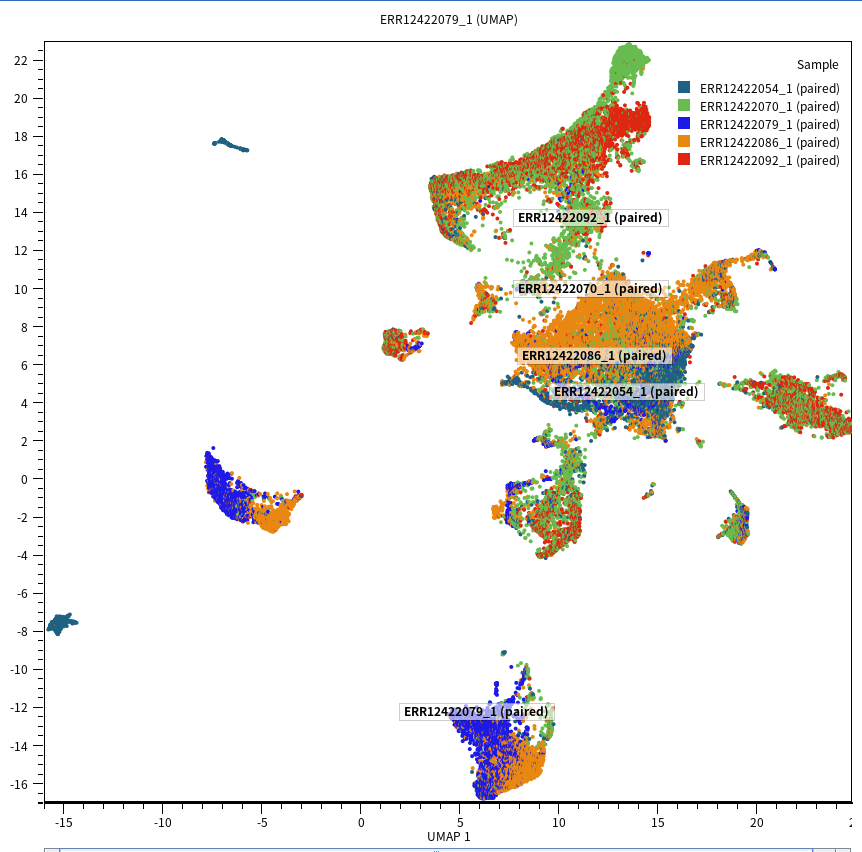
그 결과 다음과 같은 UMAP Plot이 등장했습니다. 제가 아직 파라미터 튜닝 경험이 부족해서 좀 UMAP이 이상하더라도 양해 부탁드리겠습니다.

오른쪽에 있는 coloring and highlighting 바를 활용해서 배치별로도 색깔을 구분해보았는데요. 배치끼리 비교적 잘 섞여 있는 것으로 보아 배치보정도 비교적 잘 된 것 같습니다. 참고로, 54와 70으로 끝나는게 COVID-19 샘플, 79와 92로 끝나는게 LRTD 샘플, 86으로 끝나는게 No- LRTD 샘플입니다(샘플들을 조건별로 묶을 걸 그랬네요).
이제 유사한 유전자 발현 프로파일을 갖는 세포들을 하나의 집단으로 묶는, 그렇게 군집(클러스터)을 파악하는 클러스터링을 진행해보겠습니다. 그리고 해당 클러스터들을 위의 UMAP plot에 표시해보도록 하겠습니다. 군집을 파악하는 이유는 비슷한 유전자 발현 패턴을 보이는 것끼리 묶어 특정 세포 유형으로 간주하기 위함입니다. 그렇게 특정 세포 유형에서 어떤 유전자 발현 패턴이 나타나는지 추정하는 것이 scRNA-seq의 목적이라고 볼 수 있겠습니다. 따라서 클러스터링 후에는 클러스터별 마커 유전자를 확인하여 해당 클러스터를 어떤 세포 유형으로 간주할지 annotation을 해줄 것입니다. 먼저 클러스터링을 하기 위해서는 tool box에서 'cell annotation > cluster single cell data'를 클릭하면 됩니다. 앞선 방식과 마찬가지로 정규화한 resdiuals 파일을 오른쪽에 옮겨줍니다.
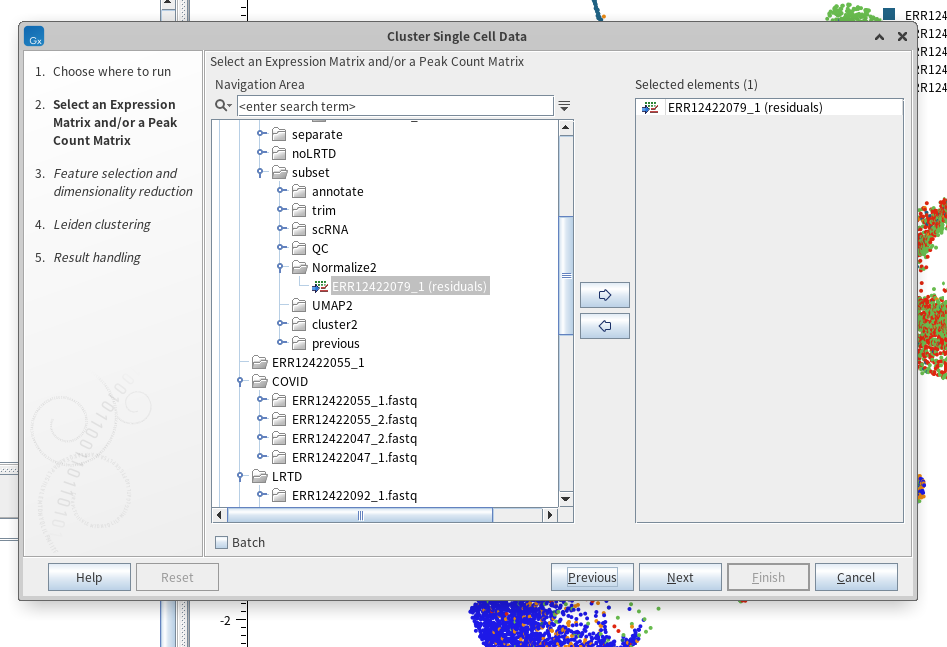
이후 저는 UMAP에서 했던 기준 값(HVG: 3000, 차원 : 20, neigborhood size : 30)대로 해서 클러스터링을 진행하였습니다. 그러면 다음과 같은 파일이 나옵니다.

파일을 눌러서 확인해보면, 해상도별로 몇 개의 클러스터가 등장했고 해당 클러스터에는 몇 개의 세포가 할당되었는지 확인할 수 있습니다. 제가 해상도(resolution)를 고정하지 않았기에 0.1부터 1.5까지 0.1 단위로 비교해볼 수 있습니다. 확인해보니 해상도 0.1로도 클러스터가 16개나 등장하기에 해상도 0.1을 기준으로 두겠습니다(배치 보정이 잘 안된건가.. 암튼 넘어가겠습니다).
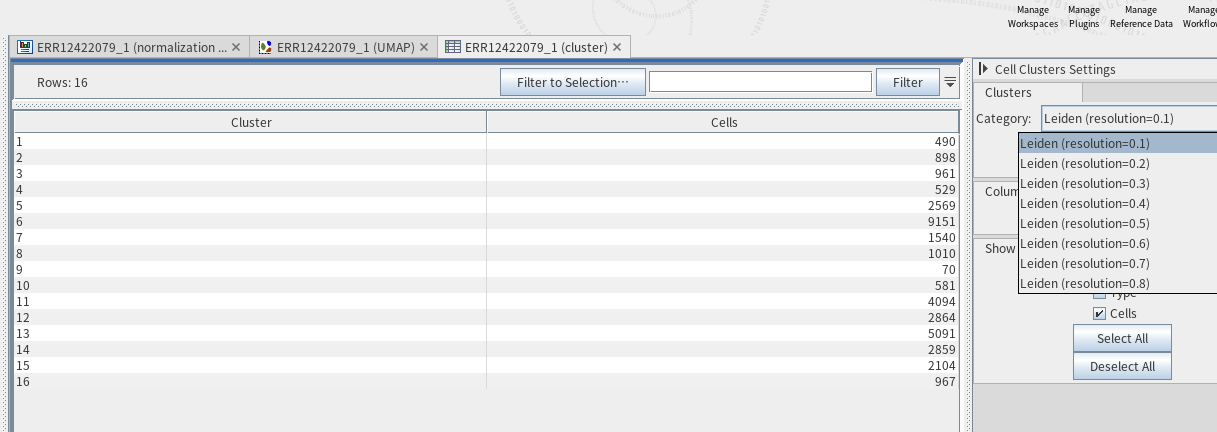
본 클러스터 결과를 앞서 구축한 UMAP plot에 표시해보도록 하겠습니다. 방법은 간단합니다. UMAP 파일을 켜고 오른쪽에 'Clusters'라고 쓰인 바에다가 cluster 결과로 나온 파일을 드래그해서 넣으면 됩니다.
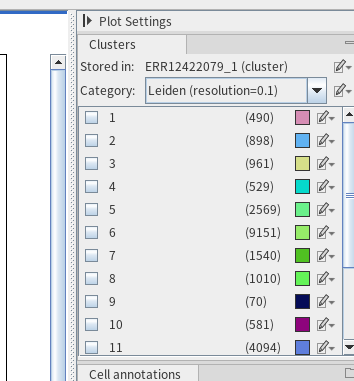
그랬더니 해상도 0.1을 기준으로 아래와 같이 16개의 클러스터가 UMAP 공간에 구분되어 표현되었습니다.

여기서 특정 클러스터 번호에 체크표시를 누르면 해당 클러스터로 분류된 세포가 강조되어 표시됩니다. 다음과 같이 말이죠.

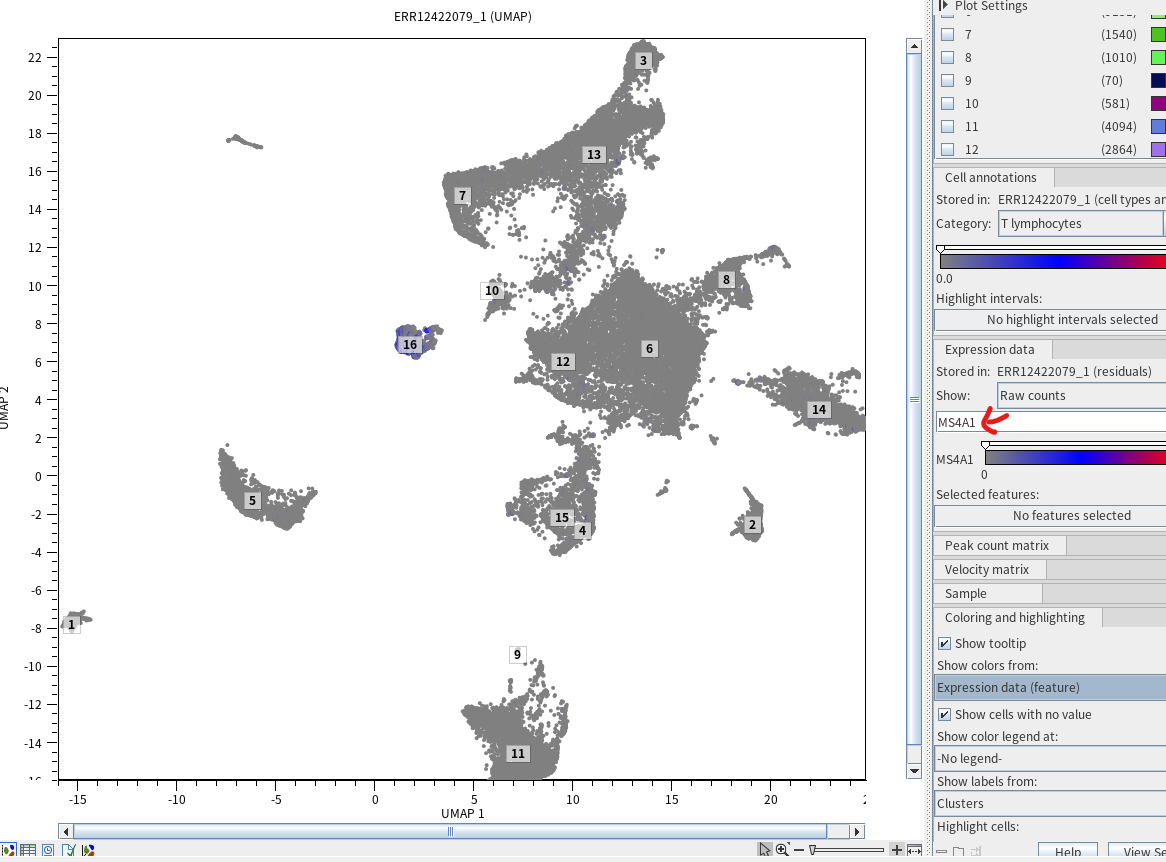
또한, 해당 UMAP 공간에서 특정 유전자를 높게 발현하는 세포 분포도 확인할 수 있는데요. 우측에 있는 Expression Data에서 다음과 같이 'MS4A1' 이렇게 특정 유전자 이름을 검색하면 됩니다. 그러면 아래와 같이 해당 유전자를 높게 발현하는 세포의 분포를 확인할 수 있습니다. B세포 마커 유전자인 MS4A1 유전자를 높게 발현하는 16번 클러스터는 B세포 유형으로 annotation할 수 있을 듯합니다. 물론 다른 마커들도 비교해봐야겠죠.
이렇게 각 면역 세포 유형의 여러 마커 유전자들의 분포를 확인하며 각 클러스터의 세포 유형을 직접 하나하나씩 annotation할 수 있습니다. 물론, 자동 annotation도 가능합니다. tool box에서 'cell type classification -> predict cell types'를 클릭하면 됩니다. 그리고나서 정규화된 데이터를 오른쪽으로 옮겨줍니다.
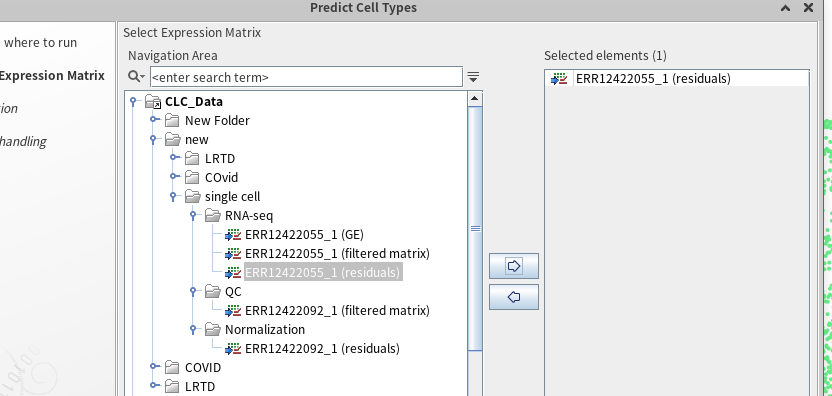
그러면 아래와 같은 화면이 등장하는데요. 앞서 다운받았던 cell_type_classifier 파일을 불러와주면 됩니다. 저는 혈액에서 얻은 샘플을 활용한 만큼 Tissue type은 따로 지정해주지 않았는데요. 특정 조직에서 얻은 샘플일 경우 어떤 조직(ex. brain, liver)에서 얻었는지 지정하면 더 신뢰도 높은 annotation 결과를 얻을 수 있을 듯 합니다(또한, 분류기도 그에 맞게 지정해주면 더 정확한 annotation을 얻을 수 있습니다). 그리고 나서 Clusters에는 앞서 얻은 cluster 파일을 불러와주고, category에서 제가 설정했던 해상도 0.1 값을 지정해줍니다. 그리고 파라미터도 적절하게 튜닝을 해줍니다.

그럼 아래와 같이 annotation 결과가 나옵니다. 보통 자동 annotation은 각 클러스터별로 세포 유형을 할당하는데 여기선 그렇지 않습니다. CLC GWB의 Single Cell Module에서는 각 클러스터별로 세포 유형을 annotate하지 않고, 개별 세포에 대해 세포 유형을 annotate합니다. 계산량이 꽤 클 것 같은데 생각보다 금방 하더군요. 그렇게 각 클러스터에 어떤 세포 유형으로 할당된 세포들이 많이 존재하는지 확인할 수 있습니다.

말초혈(PMBC)이 아닌 전혈(blood)이다 보니 이렇게 나오는 듯합니다. 또한, 분류기를 돌릴 때 조직을 지정해주지 않아서 그런지 쌩뚱맞은 세포 예측도 등장했습니다. 분류기 중에 면역세포 분류기도 따로 있다고 하는데, 그건 따로 서버에서 불러와야할 거 같아 어려움이 있어 생략했습니다. 그럼에도 연습삼아 하는 것이니 그나마 많이 예측된 T 림프구(T lymphocytes)를 대상으로 조건 별 기능 차이를 비교해보겠습니다. 참고로 위의 결과는 high confidence 예측 결과이고, 'all'로 하면 아래와 같이 T 림프구가 3천개 쯤으로 예측된 것으로 나옵니다.

우선 UMAP 공간의 우측 annotation 바에 'predict cell types' 툴로부터 얻은 'annotations'로 끝나는 파일을 드래그해서 가져옵니다.

먼저 T lymphocytes로 예측된 세포들이 어떤 클러스터에서 주로 나타나는지 확인해보았습니다. 그 결과 5번과 14번 클러스터에서 T 세포로 예측된 세포들이 주로 등장하더군요.
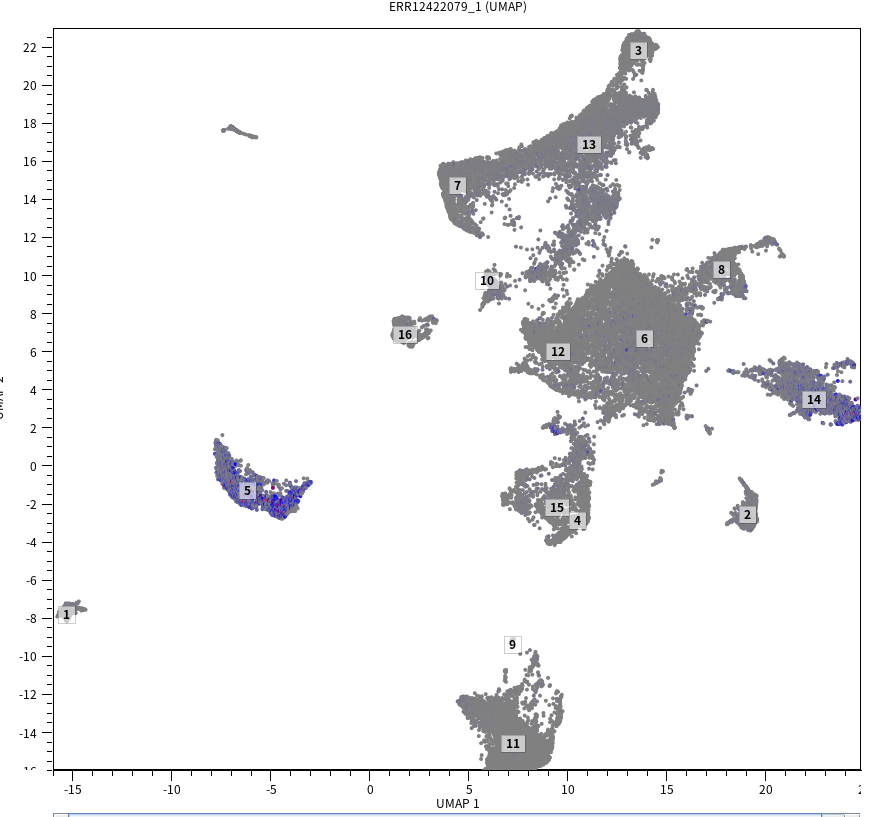
두 클러스터(5, 14)가 CD4+T세포랑 CD8+T세포로 구분되는 것인지 마커 유전자(CD4, CD8A)로 확인해보았는데요. 두 클러스터에서 골고루 나타나는 것을 보아 CD4+ T cell과 CD8+ T cell로 구분되는 건 아닌 듯 합니다.

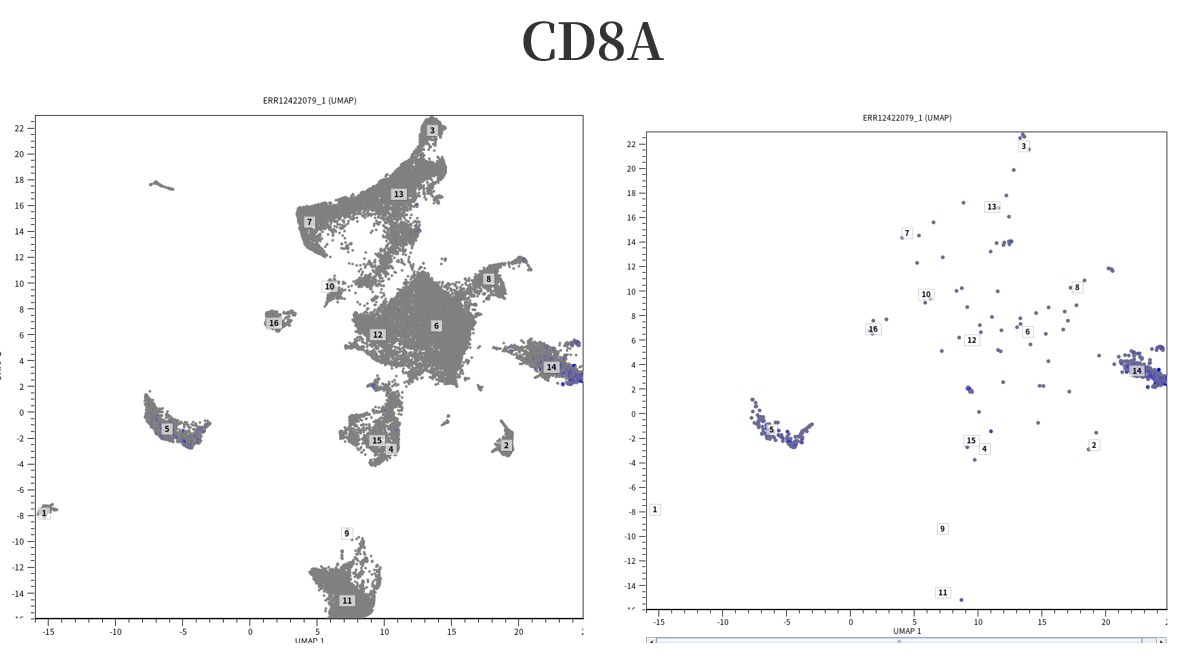
T세포는 크게 조력T세포나 조절T세포로 역할을 할 수 있는 CD4+ T 세포(CD8 -), 세포독성 T세포인 CD8+ T 세포(CD4 -)로 구분되기에 이렇게 구분이 안 되면 다운스트림 분석이 어렵습니다. 그래도 활용 예시를 보여드리는 것이니 5번 클러스터와 14번 클러스터를 T세포 상위 범주로 간주하고 다운스트림 분석을 진행해보도록 하겠습니다(원래 각 클러스터 유형에 대해 annotation을 진행해야 하지만 시간 관계상 T세포만 보는 것으로 하겠습니다). 우선 5번과 14번 클러스터 각각을 서브셋으로 따로 저장했습니다. 서브셋 파일을 만들기 위해선 아래와 같이 연필 그림 있는 부분을 클릭하고 'Creat subset'을 눌러주면 됩니다.
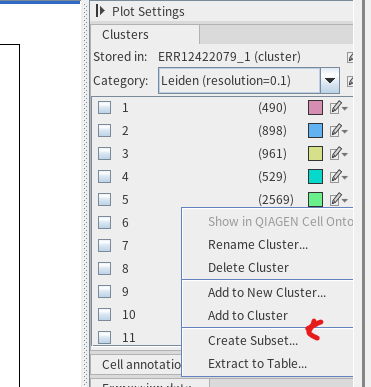
그럼 다음과 같은 파일들이 등장합니다. 2569개 세포가 있는 게 5번 클러스터, 2859번 세포가 있는 게 14번 클러스터입니다.

서브셋을 하고 파일들을 확인해보았는데요. 5번 클러스터에는 'No-LRTD인 정상환자 샘플(ERR12422086)'과 'COVID-19에 음성인 LRTD 환자 샘플(ERR12422079)'의 세포들이 주로 등장하고, 14번 클러스터에는 'LRTD 환자의 샘플(ERR12422092)'과 'COVID-19 양성 환자 샘플(ERR12422070)'의 세포들이 주로 등장했습니다. 참고로 앞서 말했듯 저는 2명의 COVID-19 환자, 2명의 LRTD 환자, 1명의 No-LRTD 환자(정상 환자) 총 5명의 샘플을 병합하였습니다.

여기서 궁금해졌습니다. T 세포로 예측된 5번째 클러스터에서 LRTD 환자의 세포들(1525개)과 NO-LRTD 환자의 세포들(924개) 간 기능적 차이가 나타나는지, 그리고 T세포로 예측된 14번째 클러스터에서 LRTD 환자의 세포들(1298개)과 COVID-19 환자의 세포들(1469) 간 기능적 차이가 나타나는지 말이죠. 이를 비교하기 위해 DEG(differental expression genes) 분석을 진행해보도록 하겠습니다. tool box의 expression analysis > create expression plot을 클릭해줍니다. 그리고 서브셋으로 얻은 정규화 파일을 옮겨줍니다(5번 클러스터와 12번 클러스터 각각 두번 진행했습니다).
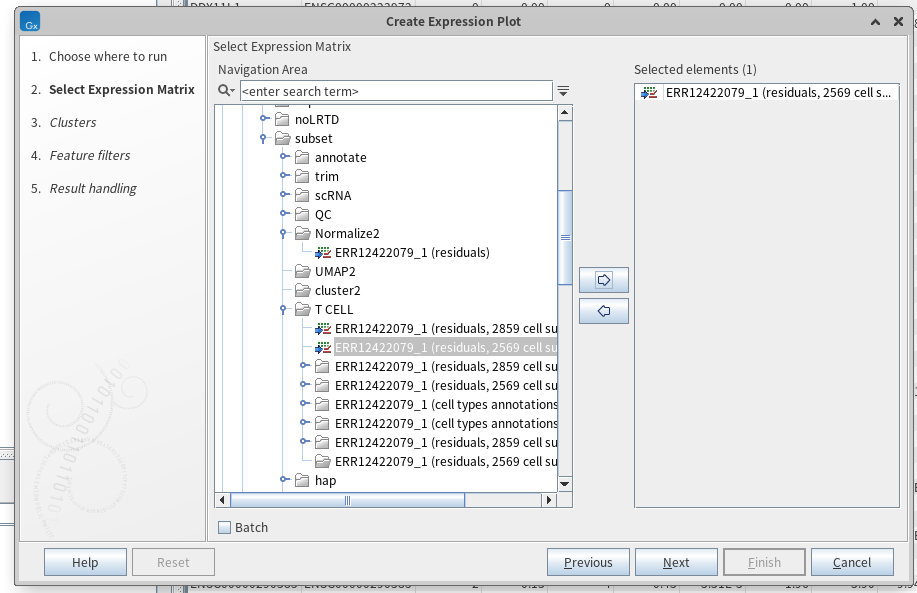
그리고 서브셋으로 얻은 cluster 파일, annotation 파일, 해상도, 비교할 샘플 등을 지정해줍니다.
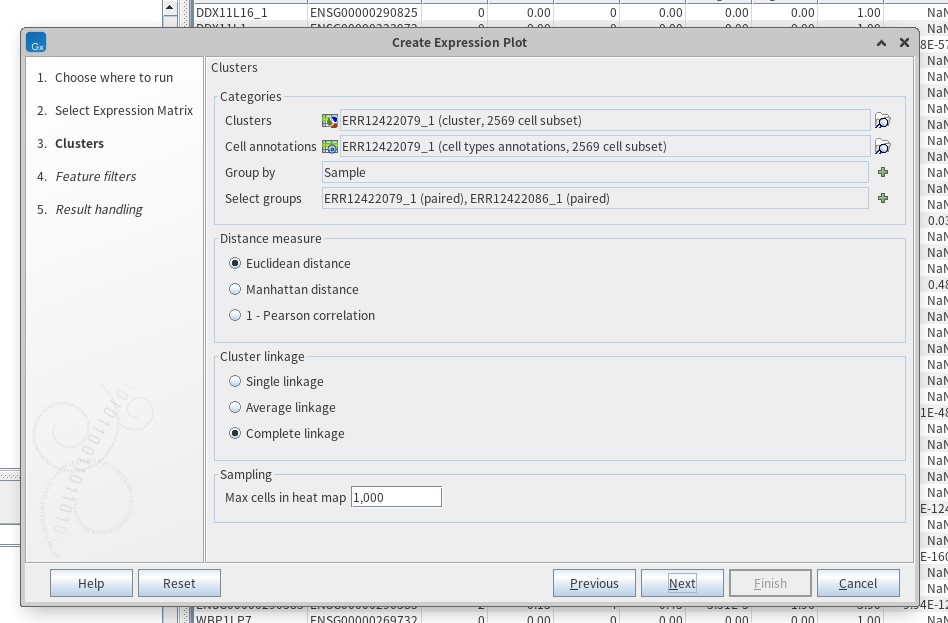
그럼 다음과 같이 두 샘플을 비교하는 Dot Plot, Heat map, Violin plot 파일이 나옵니다.

참고로 이는 5번 클러스터 대상이고, 앞서 말했듯 79로 끝나는 게 LRTD 환자, 86으로 끝나는 게 No-LRTD 환자 샘플입니다. 먼저 Dot plot 파일과 Heap 맵 파일을 열어보겠습니다.

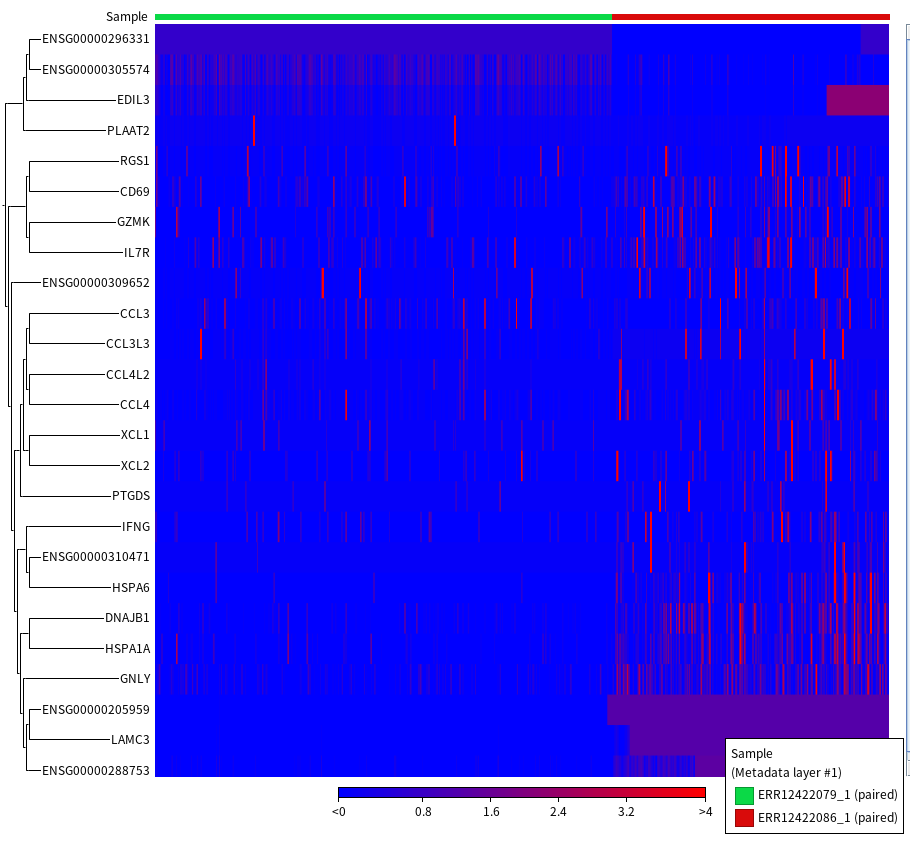
결과적으로, NK 세포나 세포독성T세포(CD8+ T 세포)에서 발현된다고 알려진 GNLY 유전자, 결핍되면 면역력 저하가 나타날 수 있는 IL7R(IL-7 수용체) 유전자 등이 No-LRTD 환자(86_1)에서 LRTD 환자(79_1)에 비해 높게 나타남을 확인할 수 있었습니다.

하나의 샘플을 사용한 거기도 하고 제대로 분석을 했는지도 모르겠지만, 이러한 결과에 대해 챗GPT는 어떻게 해석하는지 물어봤습니다. 결과는 다음과 같았습니다.


요약하면 No-LRTD 환자에서의 GNLY와 IL7R 유전자의 높은 발현은 세포독성 면역 여력이 남아 있는 '방어적 상태'를 암시하고, LRTD 환자에서의 해당 유전자들의 낮은 발현은 세포독성 풀 고갈 및 탈진으로 병원체의 하기도 침범 위험을 암시한다고 하는데요. 단순하게 질문한 것인 만큼 맞는지는 모르겠지만, 제가 체크할 수 있는 것도 아니니 넘어가도록 하겠습니다. 그래도 세포독성T세포가 기능을 잘해야 병원체의 침입을 제대로 막을 수 있는 건 확실하고, 보통 severity가 높은 조건에서 면역세포의 기능 저하가 나타났던 것 같습니다. 다음으로 14번 클러스터도 COVID-19 양성 환자 샘플(ERR12422070)과 COVID-19 음성 LRTD 환자(ERR12422092) 샘플 간에 비교를 해보았는데요. 해당 클러스터에서의 두 샘플 간의 유전자 발현 차이는 크게 나타나지 않았습니다.
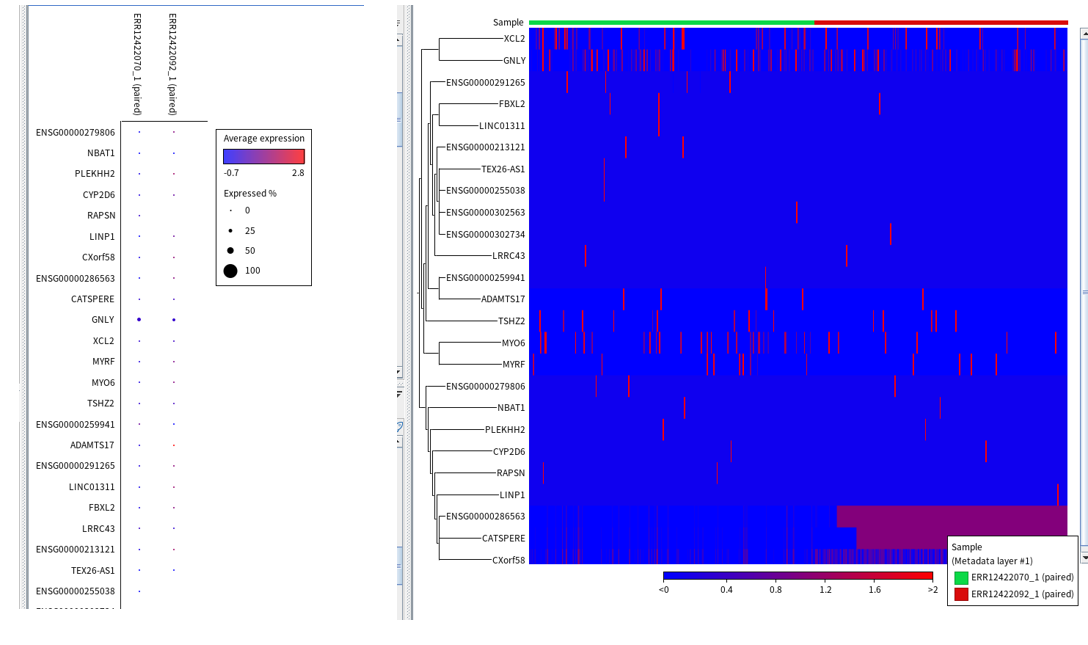
좀 더 확인해보니까 5번 클러스터에 비해 14번 클러스터는 적혈구와 관련된 마커 유전자들이 높게 발현되더군요. 분류에 오차가 있었던 것 같습니다.

그 외에도 'Differental Expression for single cell', Velocity analysis 등 다운스트림 분석을 진행할 수 있는 툴들이 여럿 존재합니다. 또한, 파일 불러오기부터 QC, 정규화, 차원축소, UMAP 표현, 클러스터링, cell type annotation 그리고 DEG 분석과 같은 여러 다운스트림 분석을 한번에 진행할 수 있는 워크플로우가 CLC Genomics Workbench에 존재합니다. tool box에서 workflows 메뉴를 클릭한 다음에 거기서 single cell workflows > from reads > expression analysis from reads를 클릭하면 됩니다.
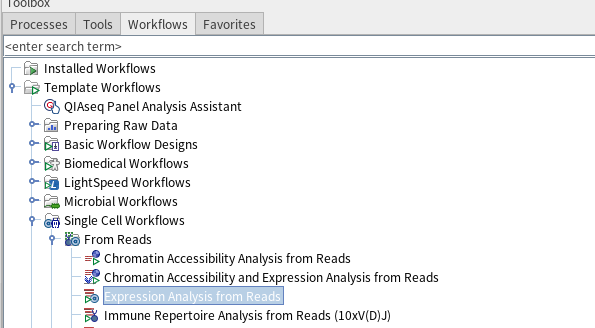
그런 다음 임포트한 데이터 샘플, 참조 서열, 기준 값 등을 몇 번 지정해주고 배치를 고려하도록 설정하고 나면 개별세포 전사체 분석이 바로 진행됩니다. 분석이 완료가 되면, 다음과 같은 파일들이 나옵니다.

앞서 봤던 리포트들도 각각 파일들로 생성해주니 확인해보셔도 좋습니다.
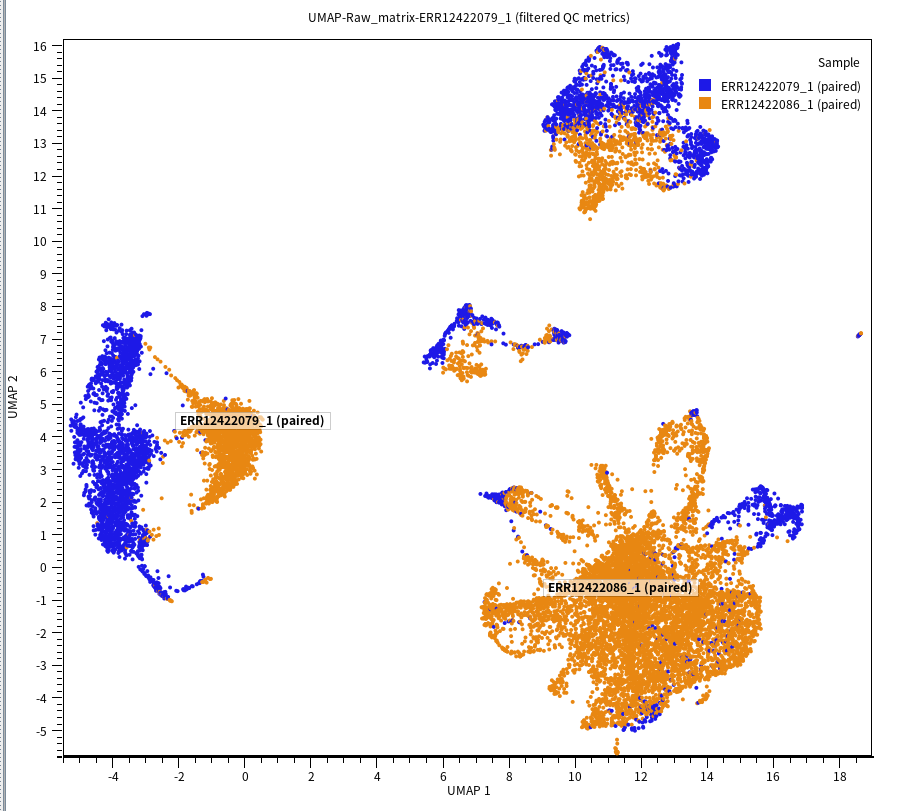
UMAP Plot은 다음과 같이 나왔는데요(해상도 0.2 기준). 배치가 두개 밖에 없는 걸 보니 제가 worflow 돌리기 위해 input 파일을 넣는 과정에서 실수를 한 것 같습니다. 배치 보정도 더 필요해보이네요.

역시 scRNA-seq 분석 쉽지 않네요. 그래도 CLC Genomics Workbench 덕분에 다양한 시행착오들을 간편하게 경험해볼 수 있었습니다. 일주일이 지난 관계로 분석은 여기까지만 해보도록 하겠습니다. 제가 사용한 single cell module 방법은 빙산의 일각에 불과합니다. CLC Genomics Workbench를 사용하면 단일 세포 전사체 분석(scRNA-seq)은 물론 오픈 크로마틴 분석(ATAC-seq), 면역 레퍼토리(TCR, BCR) 분석, 공간전사체 분석 등 멀티오믹스 분석이 가능합니다.
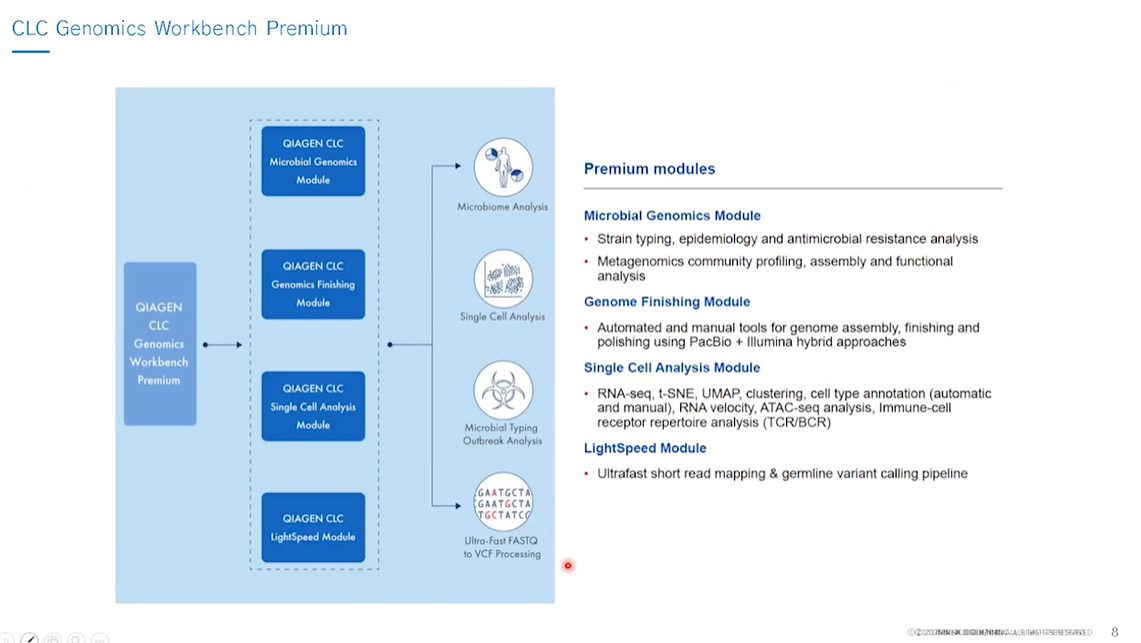
무엇보다 CLC Genomics Workbench는 '미생물 유전체 및 마이크로바이옴 분석'에 주로 활용된다고 하는데요. 이에 대한 설명은 IncoEDU 튜토리얼 강의 중 'CLC Genomics Workbench Premium 튜토리얼 - MGM 편'을 참고하시길 바랍니다. 또한, incoEDU에서 볼 수 있는 '인코세미나 61회'에서는 자주 사용하는, 그리고 최근 업데이트된 마이크로바이옴 분석 기법들을 소개해주고 있으니 본 영상도 보시면 도움이 되실 것 같습니다.
생물정보로 가는 가장 쉬운 길 | incoEDU
인코에듀는 인실리코젠만의 생물정보 교육 콘텐츠를 제공합니다. 생물정보 분야에서 활용하는 분석 솔루션의 활용법과 다양한 지식을 얻고, 생물정보에 쉽게 다가갈 수 있는 교육을 경험하세
edu.insilicogen.com
앞서 분석이 루틴할 경우 표준 Workflow를 활용하면 한번에 분석을 진행할 수 있다고 했죠. Workflow를 직접 제작할 수도 있습니다. 인코렌탈 서비스를 이용할 경우 Workflow를 만들다 어려움이 생길 때 인실리코젠 직원분께 여쭤보면 알려주실 수 있다5)고 하니 참고바랍니다.

종합적으로, CLC Genomics Workbench를 잠깐 사용해본 후기를 적어보자면, 역시 기술이 좋으면 분석이 편해지는 그런 느낌이었습니다. 코딩을 어렵게 하지 않고도 분석을 진행할 수 있게 툴들이 미리 구축해두어 있었고, 무엇보다 GUI(Graphic User Interface) 기반으로 버튼만 누르면 되었니까요. 또한, 툴들을 연결한 워크플로우까지 구축해두었으니 몇번의 클릭만으로도 한 번에 분석을 진행할 수 있는 게 좋았습니다. 물론, 이게 가격 부담이 있죠. 그럼에도 단기간만 활용할 경우 혹은 컴퓨터 용량의 한계가 있거나 분석 환경을 구축하는 데에도 어려움이 있을 경우에 인코렌탈 서비스를 이용하는게 나쁘지 않은 선택이 될 것 같습니다.

무엇보다 '인코렌탈(incoRENTAL)'은 분석 환경을 미리 구축해둔 클라우드 서버를 제공해줍니다. 이렇듯 서버를 이용하다보니 긴 시간 분석이 필요할 경우 잠자기 전에 해당 서버에 분석을 돌려놓고 일어나서 다시 원격 재접속하면 분석이 완료되어 있더군요. 물론, 단 기간도 아주 저렴한 가격은 아니지만 쓰기에 따라 활용도가 굉장히 좋은 만큼 연구에 있어 더 큰 가치를 효율적으로 창출하고자한다면 괜찮은 투자가 될 것 같습니다. 제가 아직 scRNA-seq 분석에 대해 익숙하지 않고 나머지에 대해선 더 몰라서 제대로 활용해보지 못한 것 같아 아쉬움이 남긴 합니다. 그래도 덕분에 생물정보학 분석에 활용할 수 있는 여러가지 툴들을 가볍게 접해볼 수 있었던 것 같습니다. 그럼 이것으로 인코렌탈 소개 글을 마치도록 하겠습니다. 감사합니다!
인코렌탈 | (주)인실리코젠-생물정보 전문기업
요청하신 시험판 관련 문의는 정상 접수되었습니다. 연동된 계정으로 영업일 기준 2~3일 내 답변해드리도록 하겠습니다.
insilicogen.com
- 본 글은 일정 수수료를 받고 일주일 간 인코렌탈 서비스를 체험하며 작성한 솔직 후기임을 밝힙니다 -

참고자료
1) Nyirenda, J., Hardy, O.M., Silva Filho, J.D. et al. Spatially resolved single-cell atlas unveils a distinct cellular signature of fatal lung COVID-19 in a Malawian population. Nat Med 30, 3765–3777 (2024).
2) Incoedu, CLC GENOMICS WORKBENCH 튜토리얼 PART1, 'Trim Reads', URL : https://edu.insilicogen.com/edu/my-class-room/6292?lecture=32
3) Ma, Y. uo. Integrating single-cell sequencing data with GWAS summary statistics reveals CD16+monocytes and memory CD8+T cells involved in severe COVID-19. (2022).
4) INCOEDU, CLC Genomics Workbench Premium 튜토리얼 - MGM 편, URL : https://edu.insilicogen.com/edu/course/143
5) 최영광, INCOEDU, [제61회 인코세미나] 주요 업데이트 안내 - CLC Genomics Workbench v25, URL : https://edu.insilico gen.com/edu/course/290