[57일차] MASC 모델을 활용한 세포 클러스터 구성 비율과 성별 간 연관성 분석
안녕하세요, 이번 글에서는 최근에 챗GPT를 활용해 진행해본 MASC 모델 활용 예제를 정리해보고자 합니다. 참고로 본 툴은 이전에 읽은 논문에서 활용된 툴로, 예제는 논문 데이터가 아닌 Seurat 튜토리얼에서 제공하는 간단한 PBMC 데이터로 가볍게 진행해보았습니다.

[55일차] 오픈 엑세스 논문 정리 :: Single-cell eQTL mapping reveals cell-type-specific genes associated with the risk
안녕하세요, 이번 글에서는 오픈 엑세스 논문을 하나 정리해보고자 합니다. 논문 제목은 'Single-cell eQTL mapping reveals cell-type-specific genes associated with the risk of gastric cancer'로, 저번 달에 cell genomics 저
tkmstudy.tistory.com
위의 글에서 설명했듯이 MASC(Mixed-effects Association testing for Single Cells)는 단일세포 데이터에서 '세포가 특정 클러스터(세포 유형)에 속할 오즈(odds)*'와 '임상 혹은 실험 변수(ex, 성별, 질환, 식습관 등)' 사이의 연관성을 평가하는 혼합효과 로지스틱 회귀 기법입니다. 이때 아래 식처럼 각 세포에 대해 특정 클러스터에 속할 확률을 종속변수로 둔 뒤, 공변량(여기선 성별)은 고정효과, 기증자는 무작위효과로 두어 같은 기증자의 세포들 간 내부 상관으로 인한 거짓양성 즉, 제1종 오류를 방지하면서 성별과 클러스터 구성 비율 간의 연관성이 유의한지 클러스터별로 분석합니다.
* 여기서 odds란 특정 세포가 해당 클러스터에 속할 확률을, 속하지 않을 확률로 나눈 비율을 의미합니다.

만약 클러스터 A에서 p값 0.05 미만에 오즈비가 1 미만 혹은 1 초과라면, 기증자가 여성 혹은 남성일 때 다른 성별에 비해 클러스터 A의 풍부도가 높게 나타난다고 추정할 수 있습니다. 이때 p값이 0.05 미만이라는 말은 귀무가설인 𝛽₁ = 0 즉, 공변량인 성별이 세포가 특정 클러스터에 속할 확률에 영향을 주지않는다는 가설을 기각할 만하다는 의미로 볼 수 있습니다. 다시 말해, 성별이 세포가 특정 클러스터에 속할 확률에 유의하게 영향을 준다는 뜻입니다. 그럼 MASC 워크플로를 정리해보면서 보다 구체적으로 설명해보겠습니다. 먼저 앞서 말했듯 Seurat에서 제공하는 데모용 3k PBMC 데이터를 다운받았으며, 본 파일을 R studio 환경에 가져왔습니다.

그전에 먼저 본 분석에 필요한 패키지들을 설치하고 불러왔습니다. MASC 모델 코드는 아래 깃허브 사이트에 있습니다.
GitHub - immunogenomics/masc: MASC: Mixed-effects Association testing for Single Cells
MASC: Mixed-effects Association testing for Single Cells - immunogenomics/masc
github.com
install.packages(c("devtools", "Seurat", "lme4", "dplyr", "tidyr"))
devtools::install_github("immunogenomics/masc")
다음으로 raw 데이터를 Seurat 객체로 만든 뒤 전처리를 해봅시다. 참고로 본 데이터는 이전에 분석을 해봤던 예제 데이터로 이전에 플롯을 그려보며 전처리 기준을 잡아두었기에 해당 설정값에 맞게 바로 코드를 돌렸습니다.
library(Seurat)
pbmc.data <- Read10X(data.dir = "hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 400 & nFeature_RNA < 4000 & percent.mt < 20)
pbmc <- NormalizeData(
object = pbmc,
scale.factor = 10000,
normalization.method = "LogNormalize"
)
pbmc <- FindVariableFeatures(pbmc, nfeatures = 1000)
pbmc <- ScaleData(pbmc)
pbmc <- RunPCA(pbmc)
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
pbmc <- RunUMAP(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "umap")
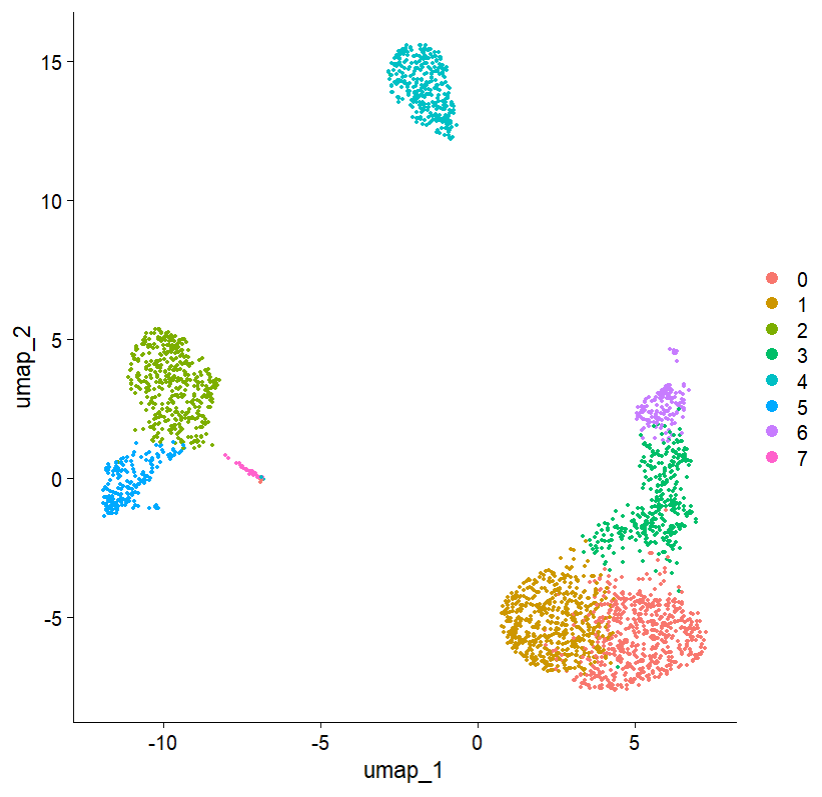
다음으로 본 예시 데이터에는 존재하지 않지만, 예시로 각 세포를 어떤 기증자로부터 얻었는지, 그리고 해당 기증자의 성별은 무엇인지에 대한 메타 데이터를 만들어보겠습니다. 저는 먼저 임의로 세포를 6명의 가상기증자로 무작위 할당을 하였습니다. 먼저 재현가능성(reproductivility)을 보장하기 위해 무작위 할당에 대한 시드를 고정하였으며, 가상 기증자를 6명으로 아래와 같이 입력했습니다.
set.seed(123)
n.donors <- 6
메타데이터를 만들기 전에 기존에 메타데이터가 담긴 seurat 객체의 @meta.data 슬롯을 'meta'라는 변수에 할당해보겠습니다.
meta <- pbmc@meta.data
View(meta)
참고로 메타데이터에는 현재 각 세포 바코드를 라벨(label)로 하여 총 RNA 수, 총 유전자 수, 미토콘드리아 유전자 비율, 그 세포가 속한 클러스터 번호 등이 적혀있습니다.

이제 메타 데이터에 6명의 기증자 ID 정보를 추가해보겠습니다. 아래 코드는 6명의 기증자를 paste0 함수를 이용해 1부터 6(n.donors)까지의 정수 벡터의 각 숫자 앞에 "D"를 붙여 c("D1", "D2", "D3", "D4", "D5", "D6")라는 문자열 벡터를 생성하고, sample 함수를 이용해 각 셀에 기증자 ID를 할당하는 코드입니다. 이때, sample함수에서는 D1 ~ D6에서 nrow(meta) 즉, Seurat 객체에 포함된 전체 세포의 개수만큼 샘플을 무작위로 추출하였고, replace =TRUE를 통해 복원 추출을 허용하여 D1~D6 중 하나를 전체 셀의 개수만큼 반복해서 랜덤하게 뽑았습니다(같은 기증자 ID를 여럿 할당).
meta$donor <- factor(
sample(
paste0("D", 1:n.donors),
nrow(meta),
replace = TRUE
)
)
View(meta)
맨 오른쪽에 각 세포를 얻은 가상의 기증자 ID가 적혀있는 donor 열이 추가되었음을 확인할 수 있습니다.
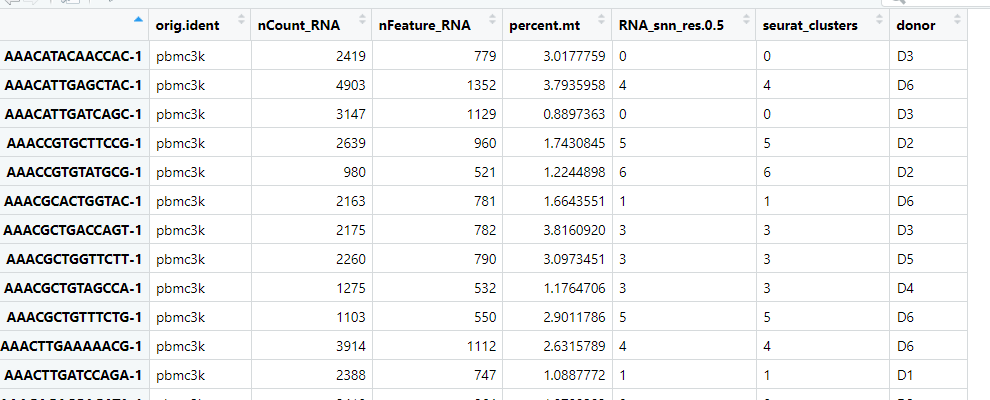
다음으로 기증자 단위로 성별을 할당한 후 meta의 새로운 열로 추가해보겠습니다. 먼저 앞선 방식과 마찬가지로 sample 함수를 이용해 'Male' 또는 'Female' 중에서 중복을 허용하여 n.donors의 수(6명)만큼 무작위로 뽑고*, setNames(vectors, names) 함수를 통해 무작위로 뽑은 성별 벡터에 각 요소에 대응되는 이름(기증자 ID 명 벡터)을 붙입니다.
donor_sex <- setNames(sample(c("Male","Female"), n.donors, replace = TRUE)),
paste0("D", 1:n.donors))
donor_sex
-> D1 D2 D3 D4 D5 D6
"Male" "Female" "Male" "Female" "Female" "Female"
# * 'sample(c("Male","Female"), n.donors, replace = TRUE)'만 실행하면?
-> Ex. "Male" "Female" "Male" "Female" "Female" "Female"
위에 적었듯 donor_sex 변수는 이름(name)이 D1~D6인 벡터(batch name)이고, 값(value)은 Male 또는 Female입니다. 따라서 donor_sex[meta$donor]는 각 세포마다 해당 세포의 기증자 ID에 대응하는 성별을 벡터 형태로 반환합니다. 다음과 같이 말이죠.

이렇게 얻은 성별 벡터를 factor 함수를 사용해 범주형으로 변환하면서 명시적으로 레벨을 c("Female", "Male") 순서로 지정합니다.
meta$sex <- factor(donor_sex[meta$donor], levels = c("Female","Male"))
결과적으로, level=0일 때 Female(여성), level = 1일 때 Male(남성)이 되었는데, 이렇게 레벨 순서를 지정하면 통계 모델링시 Female이 기준(reference)이 되고, 'Male의 Female 대비 차이'가 나타나게끔 설정되는 효과가 있습니다. 즉, 향후 회귀분석에서 성별(sex)을 공변랑으로 넣었을 때 R은 자동으로 Female을 기준으로 두고 Male의 회귀계수를 계산하게 됩니다. 이에 대해선 뒤에서 자세히 설명하도록 하겠습니다. 암튼 이렇게 해서 각 세포마다 기증자의 성별이 기증자 ID에 맞게 factor로 저장되었습니다. 다시한번 meta를 확인해보니 오른쪽에 성별 정보가 담긴 열이 추가되었네요.

이제 수정된 메타 데이터를 Seurat 객체 'pbmc'의 @meta.data 슬롯에 다시 넣겠습니다.
pbmc@meta.data <- meta
이것으로 데이터 준비가 완료되었는데요. 본 데이터를 MASC 모델에 돌릴 수 있는 데이터 프레임 형태로 변환해보겠습니다. 이를 위해 앞서 생성한 meta에서 transmute 함수를 사용해 필요한 열들만 남기고 나머지는 모두 삭제해보겠습니다. 참고로, transmute 함수는 dplyr 패키지가 제공하는 함수로서 새로운 열 이름을 만들어서 반환하되 기존 열 중에서 새로운 열 이름으로 지정한 것만 남기고 나머지 열은 모두 삭제하는 함수입니다. 이때 본 코드에서는 meta |> 연산자를 사용하여 meta를 transmute 함수의 첫번째 인수로 넘겨주면서 그로부터 나온 결과를 masc.df에 저장했습니다.
library(dplyr)
masc.df <- meta |>
transmute(
cluster = factor(seurat_clusters),
donor = donor,
sex = sex,
nUMI = nCount_RNA
)
참고로 여기서 cluster는 MASC 모델의 종속변수나까 추가하고, donor는 기증자별 내부 상관을 무작위 효과로 모델링하기 위해 추가하고, sex는 여성과 남성을 비교하기 위한 설명 변수니까 추가하고, nUMI는 라이브러리 크기에 따른 편차를 보정하기 위한 고정 효과로 모델링하기 위해 추가했습니다. 그렇게 네 개 열을 뽑아 masc.df에 저장하면 결과적으로 다음과 같은 형태의 데이터 프레임이 만들어집니다.

이제 masc 패키지의 MASC 모델로 분석을 돌려보고자 합니다. 저는 아래 깃허브의 MASC 함수 코드를 가져와 MASC 함수를 사용하였습니다.
masc/R/masc.R at master · immunogenomics/masc
MASC: Mixed-effects Association testing for Single Cells - immunogenomics/masc
github.com
그렇게 함수를 만들고 나서 다음과 같이 사용했습니다. 앞서 말했듯 사용할 데이터 프레임 명, 종속변수, 설명변수, 무작위효과, 고정효과를 지정해주었습니다.
library(lme4)
res <- MASC(
data = masc.df,
cluster = masc.df$cluster,
contrast = "sex",
random_effects = "donor",
fixed_effects = "nUMI"
)
그러면 다음과 같은 결과값 데이터프레임 'res'가 나옵니다. 성별에 따라 비율 차이를 유의하게 나타내는 클러스터는 1번인 듯합니다.

참고로 여기서 오즈비는 sexMale.OR에서 확인할 수 있는데 이 값이 앞의 레벨 기준에 따라 1보다 크면 남성에서 해당 클러스터의 세포 비율이 더 높다고 볼 수 있고, OR < 1이면 여성에서 해당 클러스터의 세포 비율이 더 높다고 볼 수 있습니다. 그 이유는 아래 식에서 확인할 수 있습니다. 물론 유의한 OR이라고 판단하려면 앞서 말했듯 p값이 최소 0.05 미만이어야 하겠죠.
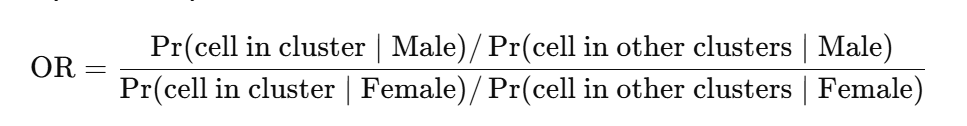
본 결과를 보기 좋게 시각화해보겠습니다. 시각화 방법은 다양하겠지만, 제가 MASC 모델을 접한 논문에서처럼 x축에 성별 오즈비(OR), 세로축에 -log10(p값), 점 하나를 각 클러스터(ex. cluster0)로 표시해 시각화하는 것이 보기 좋은 것 같습니다(아래 논문 Fig2A 참고).
Figure - PMC
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
pmc.ncbi.nlm.nih.gov
그럼 그렇게 해보겠습니다. 먼저 시각화에 필요한 패키지 'ggplot2'와 'ggrepel'를 설치하고 불러옵니다.
install.packages("ggplot2")
install.packages("ggrepel")
library(ggplot2)
library(ggrepel)
다음으로 model.pvalue로부터 -log10을 취한 값을 저장한 열을 추가하고, 기준선(ex. OR = 1, p값 = 0.05) 값도 구해둡니다.
res$negLog10P <- -log10(res$model.pvalue)
vline_or <- 1
hline_pval <- -log10(0.05) # -log10(0.05) ≈ 1.301
챗GPT가 만들어준 ggplot 함수를 이용한 시각화 코드는 다음과 같습니다. 코드를 구체적으로 살펴보면 geom_point()를 이용해 각 클러스터별 오즈비와 -log10(p-value_에 해당하는 좌표에 점을 찍고, geom_text_repel()을 이용해 cluster 이름을 각 점 옆에 표시하면서 서로 겹치지 않도록 자동 조정해줍니다(ggrepel 패키지 사용).
ggplot(res, aes(x = sexMale.OR, y = negLog10P)) +
geom_point(size = 3, color = "steelblue") +
geom_text_repel(
aes(label = cluster),
size = 5,
box.padding = 0.3,
point.padding = 0.3,
segment.color = "gray60"
)
이후 geom_vline()와 geom_hline()로 시각적으로 OR=1 기준선과 유의수준 기준선을 더해서, 해석을 쉽게 해 줍니다. 또한. labs(x= "", y="", title="")을 통해 축 제목과 그래프 제목을 지정하고, theme_minimal()으로 최소한의 테마만 사용하도록 했습니다. 이렇게 하면 다음과 같은 플롯이 등장합니다.
# 앞의 코드와 이어서
+ geom_vline(xintercept = vline_or, linetype = "dashed", color = "darkred") +
geom_hline(yintercept = hline_pval, linetype = "dashed", color = "darkred") +
labs(
x = "Odds Ratio (Male vs Female)",
y = expression(-log[10]*"(p-value)"),
title = "MASC 결과: 클러스터별 OR vs p-value"
) +
theme_minimal(base_size = 14) +
NULL
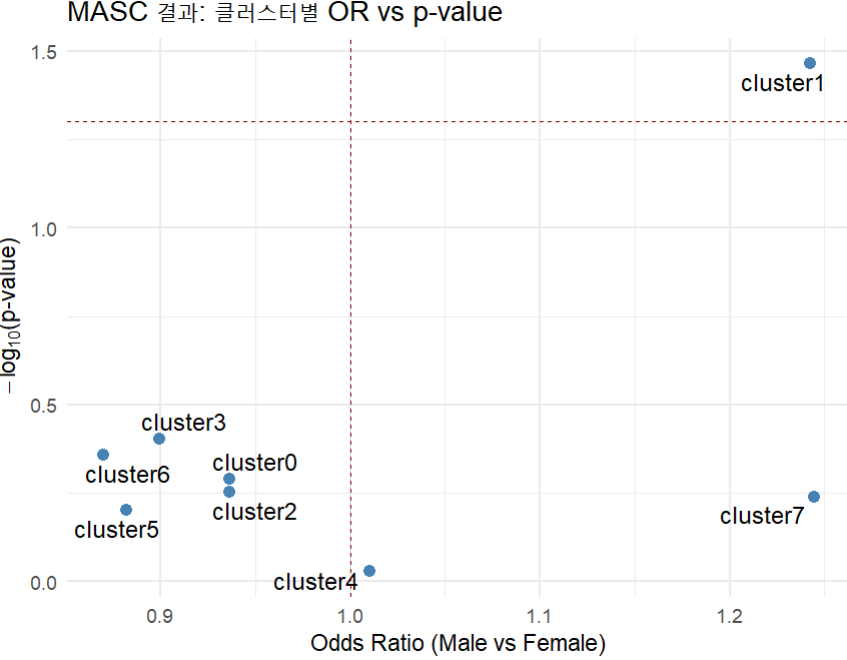
여기서 가로축(OR)에서 1을 기준으로 왼쪽은 Female 우세, 오른쪽은 Male 우세 영역이고 세로축(−log₁₀(p-value))에서 1.301 위쪽은 유의수준(p < 0.05) 영역입니다. 본 플롯을 통해 결과적으로 어떤 클러스터가 성별에 따라 유의하게(MASC p < 0.05) 앞으로(OR > 1) 혹은 뒤로(OR < 1) 치우쳤는가를 한눈에 볼 수 있습니다. 다음 툴도 튜토리얼을 챗GPT보고 만들어보도록 한 뒤 정리해보겠습니다. 감사합니다!
참고자료
1. 챗GPT