안녕하세요, 전 글에 이어서 'Single-cell eQTL mapping reveals cell-type-specific genes associated with the risk of gastric cancer' 논문 정리를 해보겠습니다.
[55일차] 오픈 엑세스 논문 정리 :: Single-cell eQTL mapping reveals cell-type-specific genes associated with the risk
안녕하세요, 이번 글에서는 오픈 엑세스 논문을 하나 정리해보고자 합니다. 논문 제목은 'Single-cell eQTL mapping reveals cell-type-specific genes associated with the risk of gastric cancer'로, 저번 달에 cell genomics 저
tkmstudy.tistory.com
앞선 내용을 짧게 설명하면, 본 연구팀은 scRNA-seq 데이터로 파악한 19개의 위 조직 세포 아형을 대상으로 세포 아형 조성 비율과 관련된 거시적인 요인들과 유전적 요인(SNP)의 영향을 조정하면서 cell-type specific eQTLs를 파악하고 그 위치의 eSNP로부터 발현이 조절되는 eGene들을 파악하였습니다. 그리고 나서 eGene에 대한 세포 아형 간 공유 eQTL 신호와 독립 eQTL 신호를 구분하였습니다.
다음으로 본 연구팀은 101명의 pairwise samples에서 각 유전자의 cell-type-specific expression levels와 실제 bulk expression levels 사이에서의 상관관계를 비교하였습니다. 그 결과, mucous pit cell과 mucous neck cell에서는 본 상관관계가 전반적으로 높게 나타났으며, artery-derived EC(endothelial cell)에선 상관관계가 상대적으로 낮게 나타났다고 합니다. 일부 유전자의 경우 세포유형별로 상관관계의 방향성이 다르게 나타난 경우도 있었는데요. mucous pit cell에서의 MUC5AC의 발현은 bulk tissue expression levels와 positive correlation을 보였으나, CD4+ T cell에서의 MUC5AC의 발현은 negative correlation을 보였다고 합니다.
그 이유에 대해 잠깐 설명하고 넘어가자면, 우선 샘플에서 차지 비율이 높은 세포 아형(약 17.4%, 19개 세포 유형 중 2위)인 mucous pit cell은 MUC5AC 유전자를 높게 발현하므로, 이 세포에서의 MUC5AC 신호가 강해질수록 bulk 시료의 MUC5AC 발현도 증가한다고 볼 수 있습니다. 반면 CD4⁺ T 세포는 MUC5AC를 거의 잡음 수준으로만 발현하므로, CD4⁺ T 세포에서 관측되는 MUC5AC 신호가 상대적으로 높다는 것은, mucous pit cell의 MUC5AC 신호가 낮아졌음을 의미합니다. 따라서 CD4⁺ T 세포의 MUC5AC 신호 증가는 bulk 수준의 MUC5AC 발현과 음의 상관관계를 보인다고 해석할 수 있습니다. 결국에 bulk(샘플 전체) 수준에서 특정 유전자의 발현 수준을 결정하는 것은 샘플 내 차지 비율이 높은, 그러면서도 특이적인 유전자 발현 패턴을 갖는 세포 유형이니까요(물론 샘플 구성이 다양하면 꼭 그렇지는 않을 수 있습니다).
참고로 본 분석에서 연구팀은 기증자별로 특정 유전자의 '세포-특이 발현량'과 '실제 bulk 발현량'을 1대1로 매칭한 뒤 pearson 상관 분석을 적용해 유전자가 어느 세포 유형에서 bulk 발현을 주도하는지(양의 R) 혹은 반대 방향으로 기여하는지(음의 R)를 판별했습니다. 여기서 피어슨 상관 분석을 적용해서 계산하는 '피어슨 상관 계수'는 두 특징 사이의 선형 상관관계의 세기이자 [-1, +1] 구간의 값으로, 여기서 '선형'은 두 특징의 상관관계가 얼마나 강한지를 하나의 직선으로 서술할 수 있다는 의미입니다. 이렇듯 101개의 scRNA-seq–bulk pairwise 샘플에서 세포 유형별 평균 발현량(pseudo-bulk)과 샘플 전체에서의 평균 발현량(bulk-seq)을 유전자 단위로 일일이 상관분석한 이유는 한 사람이 가진 적은 양의 세포특이 유전자 발현 정보가 실제 조직 전체(bulk)에서 관측되는 신호와 일관되는지를 확인해 단일-세포 데이터의 정량적 신뢰도를 교차 검증하기 위함이라고 볼 수 있겠습니다. 보다 구체적으로 설명하자면 scRNA-seq은 한 샘플을 수천~수만 개의 cell barcode로 쪼개서 관측하기 때문에 어떤 유전자가 이 샘플에서 얼만큼 발현되는지를 정량하기에는 세포 당 전사체 총량이 적을 수 있다는 한계가 있습니다. 이러한 한계는 0으로 측정된 값이 진짜 무발현인지 아니면 단순히 잡히지 않은 발현인지 구별하기 어려운 드롭아웃 현상으로도 이어질 수 있습니다. 반면 동일 샘플에서 얻은 bulk RNA-seq은 단일세포보다 훨씬 깊은 read coverage를 갖기 때문에 샘플 수준 평균 발현이라는 관점에서 신뢰도가 높습니다. 따라서 본 연구팀은 scRNA-seq을 통해 단일세포수준에서 유전자 발현 프로파일을 확인한 뒤 bulk RNA-seq으로 해당 유전자의 발현을 read coverage가 높은 샘플 전체 수준에서 한번 더 확인하고, 두 가지 수준의 발현량의 상관관계를 비교한 것이라고 할 수 있겠습니다.
문제는 조직 내 희귀 세포형에서는 특정 유전자가 세포 내부에서 매우 강하게 발현되더라도, 해당 세포가 조직 전체에서 차지하는 비율이 극히 작기에 같은 시료의 bulk RNA-seq에서는 그 유전자의 총 신호가 희석되어 낮게 나타날 수 있다는 것입니다. 실제로 101개의 bulk-scRNA-seq 짝 샘플을 분석한 결과, 세포 유형별 평균 발현과 bulk 발현 사이의 상관계수는 세포 비율에 따라 다르게 나타났다고 하는데요. 구체적으로 비율이 높은 mucous neck cell과 mucuos pit cell은 bulk 발현과 뚜렷한 양의 상관을 보였지만, 드물게 나타난 artery-derived endothelial cell은 평균 r = 0.05로 상관관계가 거의 없었다고 합니다. 물론 이때 낮거나 음(-)으로 나타난 상관계수는 헤당 세포형이 bulk 발현에 크게 기여하지 않는다는 뜻일 뿐, 그 세포 내부에서의 발현 강도를 부정하는 지표는 아닙니다.
세포 조성에 따른 편향을 보정하기 위해 연구팀은 기증자별 bulk RNA-seq 시료(기증자당 1개)에서 allele-specific expression(ASE)을 계산하고 이를 단일세포 eQTL 결과와 비교하였습니다. 이때 ASE는 이배체(두 세트의 유전체를 지닌) 생물에서 한 유전자의 두 대립형(alleles) 가운데 한쪽이 다른쪽보다 더 많이 전사되어 RNA 수준에서 발현 불균형이 나타나는 현상을 말하는데요. 연구팀은 이러한 ASE 신호를 확보하기 위해 우선 기증자의 bulk RNA-seq 리드를 STAR(v2.6.1a)를 이용해 GRCh37 참조 유전체에 정렬(alignment)한 뒤, WASP 파이프라인을 적용해 대립유전자 매핑 편향(allele-specific mapping bias)을 교정한 BAM 파일을 생성했습니다. 이때 매핑 편향을 바로잡지 않으면, 참조 대립유전자(예: A)를 포함한 리드는 우선적으로 매핑되고, 대체 대립유전자(예: G)가 들어 있는 리드는 탈락하거나 낮은 mapping quality를 받아 실제 발현 차이를 과소추정하게 될 수 있습니다. 따라서 WASP는 각 리드에서 SNP를 가린 뒤(reference-masked remap) 재정렬을 수행하고, 재정렬 결과가 처음과 다르면 해당 리드를 삭제해 두 대립유전자에 동일한 정렬 기회를 보장했습니다. 이후 중복 리드를 제거하고 CIGAR·MD 태그*를 유지하여, phASER가 read 단위로 하플로타입(H1/H2)을 정확히 할당할 수 있도록 했습니다. 참고로 여기서 하플로타입(haplotype)은 한 사람의 두 염색체 가운데 같은 부모로부터 이어받은 한쪽 염색체에 연속해서 존재하는 대립유전자들의 조합을 가리키며, H1과 H2는 각각 '부모 1(ex. 아빠)에서 온 염색체'와 '부모 2(ex. 엄마)에서 온 염색체'에 위치한 변이 세트를 뜻합니다.
* CIGAR와 MD 태그는 각 리드가 참조 서열에 어떻게 매핑됐는지(ex. 삽입, 삭제, 불일치 위치)와 변이 지점에서 실제로 관찰된 염기를 함께 기록해, 단일 리드만으로도 염기 수준의 정렬 + 변이 정보를 모두 제공합니다. phASER는 이 두 태그를 읽어 리드가 포함한 대립유전자를 파악한 뒤, 사전에 phase된 VCF의 0|1 또는 1|0 패턴과 일치 여부를 확인해 “변이 A와 변이 B 모두에서 H1 대립유전자를 지니므로 이 read는 H1 하플로타입에 속한다”처럼 일관된 하플로타입을 판단합니다. 이때 사전에 중복 리드를 제거해 두면 동일 DNA 조각이 여러 번 집계되는 것을 방지할 수 있어 하플로타입별 리드 수와 이후 ASE 정량이 훨씬 정확해집니다.

본 연구팀은 ASE 정보를 얻기 위해 BAM 파일 뿐만 아니라 같은 기증자의 유전체 데이터(SNP-array + imputation 데이터)를 SHAPEIT(v2.12)로 위상 결정(phasing)함으로써 VCF 파일을 얻었습니다. 본 VCF 파일은 이형접합 SNP마다 GT(genotype field)가 0|1 또는 1|0 형식으로 되어 있는데, 0은 참조 allele, 1과 2는 대체 allele을 가리키고, | 기호는 앞쪽 숫자가 하플로타입 1(H1), 뒤쪽 숫자가 하플로타입 2(H2)에 위치함을 나타냅니다(앞서 말했듯 H1과 H2는 각각 '부모 1에서 온 염색체'와 '부모 2에서 온 염색체'에 위치한 변이 세트를 뜻합니다). phASER GENE AE는 이 BAM과 VCF 파일을 읽어 'chr11:12345 A>G 변이(한 염기가 A에서 G로 바뀐 단일염기변이(SNV)*)에서 H1 = 60, H2 = 55 read”, “MUC5AC 유전자에서 H1 = 120, H2 = 80 read'처럼 기증자 X 유전자 수준의 하플로타입별 리드 수를 산출합니다. 그렇게 본 연구팀은 여러 기증자의 데이터를 통합해 하플로타입 기반 발현 행렬(haplotypic expression matrix)을 구축했고, 각 하플로타입에 매핑된 read 개수를 계산해 이를 allele-specific expression (ASE)으로 정의했습니다.
* SNV는 모든 단일염기변이이고, SNP는 그중 인구집단에서의 대체 대립유전자 빈도(보통 ≥ 1%)가 일정 수준 이상으로 유지돼 다형성(polymorphism)으로 인정된 변이(주로 생식계열 변이를 지칭)를 말합니다.
ASE는 이형접합 SNP 두 대립형 가운데 한 쪽이 과다전사되는 불균형을 이용하므로, 리드 수가 많지 않아도 이형접합 리드만 충분하면 조절 변이를 검출할 수 있습니다. 다시 말해, ASE 분석은 세포 비율에 크게 좌우되지 않으면서도 희귀 세포형-특이 조절 효과까지 포착할 수 있는 보완 지표로 작동할 수 있다고 볼 수 있겠습니다. 실제로 본 연구에서는 Bulk-eQTL 분석에서와 달리 Bulk-ASE 분석에서 epithelium-lineage cells(mucous neck cells, mucous pit cells)를 넘어 전체 세포형에 걸쳐 eQTLs의 수가 일관되게 나타남을 확인했습니다(특히, 큰 샘플 사이즈를 갖는 Bulk-GTEx 데이터셋).
다음으로 본 연구팀은 25개 위암(GC) GWAS sensitivity loci(기존 GWAS 문헌에서 보고된 lead SNP와 강한 LD(r^2 > 0.5)인 영역)를 단일세포 eQTL과 연결하기 위해, 각 세포 유형에서 얻은 lead eSNP 집합과의 중첩(overlap)을 계산하였습니다. 이때 eSNP(또는 이를 태그하는 LD SNP)가 25개의 loci 가운데 하나와 LD(r^2 > 0.5)이면 overlap으로 정의하였습니다. 또한, 세포 유형 간에 eQTL(lead eSNP 집합)이 위암 GWAS 좌위에 풍부한 정도를 비교하기 위해 카이제곱 검정으로 세포 간 빈도 차이를 검정하였습니다. 분석 결과, 상피(epithelial) 계열, 특히 mucuous neck cell과 mucous pit cell에서 eQTL과 GC GWAS loci 간 중첩 비율이 가장 높았고, 면역/섬유모세포/내피 계열에서는 현저히 낮았습니다. 이때 상피 계열에서 중첩된 대표적인 감수성 유전자(sensitiviy genes)로는 PTGER4(5p13.1)와 FRG1B(20q11.21)가 확인되었으며, 8q24.3 좌위의 PSCA는 다수의 세포 유형에서, 6p21.1 좌위의 APOBEC2는 parietal 세포에서만 발현 연관성이 나타났습니다. 본 결과들은 GWAS 위암 위험 변이가 특정 세포 유형(단일 혹은 다수)에서의 특정 유전자 발현 조절을 통해 위암 위험을 매개할 수 있다는 가능성을 시사합니다.

다음으로 본 연구팀은 각 세포 아형에 대한 GC GWAS 데이터와 eQTL 신호 간의 공위치화(co-localization) 분석을 수행하였습니다. 앞서 말했듯 GWAS 신호와 eQTL 신호가 동일한 SNP에서 공위치화된다면, 그 변이가 형질에 영향을 주는 메커니즘이 그 유전자 발현 변화를 매개하고 있을 가능성을 시사합니다. GWAS와 eQTL 신호의 공위치화를 평가할 때는 다음과 같은 순서로 진행하는데요. 먼저 동일한 유전자 주변(region of interest, 보통 ±1Mb)에 존재하는 GWAS 요약 통계(효과 크기 β_GWAS, 표준오차 SE_GWAS, 대립유전자 빈도 등)와 cell type specific eQTL 요약통계(β_eQTL, SE_eQTL, 대립유전자 빈도)를 수집한 뒤, GWAS 신호와 eQTL 신호가 동일한 변이에 의해 설명될 가능성이 높은지 통계적으로 판단합니다.
본 연구진은 위암(GC) GWAS 신호와 각 세포 유형별 cis-eQTL 신호가 동일한 인과 변이를 공유하는지 검정하기 위해 R 패키지 coloc (v 5.2.2)의 summary-statistics용 근사 베이지안(Bayes factor) 방법을 이용했습니다. 본 방법에선 우선 각 유전자에 대해 주변 ±1 Mb 구간에 속하는 SNP들의 GWAS 요약 통계와 해당 세포 유형 eQTL 요약 통계를 추출한 뒤, 두 형질(유전자 발현 / 질병)의 공유, 비공유, 독립을 가정하는 다섯 가지 가설 H0 ~ H4(PP0 - PP4)에 대해 근사 베이지안 인자(ABF)를 계산했습니다*. 여기서 coloc은 SNP별 인과 구성을 S로 정의하고, 관측 데이터 D가 주어졌을 때 다음과 같은 식으로 가설별 사후확률을 산출합니다.

* PP0는 GWAS 신호와 eQTL 신호 모두와 연관된 인과 변이가 전혀 없는 경우, PP1은 인과 변이가 eQTL 신호(형질 1)에만 존재하는 경우, PP2는 인과 변이가 GWAS 신호(형질 2)에만 존재하는 경우, PP3는 두 신호가 서로 다른 인과 변이에 의해 독립적으로 조절되는 경우, PP4는 두 신호가 동일한 인과 변이를 공유하는 경우를 뜻합니다.
여기서 PP4는 GWAS와 eQTL이 동일한 단일 인과(casual) SNP를 공유할 사후확률이며, 아래와 같이 표현됩니다. 여기서 분석은 각 유전자 x 세포 유형마다 독립적으로 수행되었고, PP4 > 0.7을 만족할 때 그 유전자-세포유형 쌍이 GC GWAS 신호와 공위치된다고 판정하였습니다.
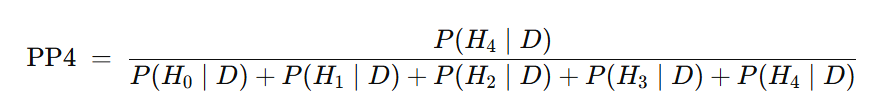
결과적으로 17개의 공위치화 신호가 위암(gastric cancer) 맥락에서 서로 다른 위 세포 조성(gastric cell types)에 걸쳐 파악되었으며, 본 결과는 Fig5A(아래 링크 참조)에서 확인할 수 있습니다. 한 예로, 4q28.1* 위치에서의 ANKRD50 유전자와 6p21.1에서의 APOBEC2 유전자가 각각 mucous neck cells와 parietal cells에 특이적으로 공위치화된다는 것을 확인할 수 있었습니다. 종합적으로, 본 결과는 단일세포 eQTL 데이터가 세포 유형 특이적 유전자 발현과 특정 형질(ex. 위암)과 연관된 유전 변이를 연결해 변이의 기능적 영향을 규명할 수 있는 잠재력을 보여준다고 할 수 있겠습니다.
* 4 / q / 2 / 8 / .1 : 염색체 번호 / 팔(p = short arm, q = long arm) / region / band(region 안에서의 굵은 줄무늬) / sub-band(banc를 더 세분화한 구간)
Figure - PMC
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
pmc.ncbi.nlm.nih.gov
다음으로 본 연구팀은 다양한 위 세포형(gastric cell types)에서 유전적으로 예측된 유전자 발현(mRNA) 수준과 GC risk 사이의 관계를 조사하기 위해 cell-type-specific TWAS를 수행하였습니다. 참고로 여기서 TWAS (Transcriptome-wide association studies)는 유전 변이(ex. SNP)에 의해 조절되는 특정 형질과 연관된 유전자(trait-associated genes)를 발굴하는 유전자-우선순위화(gene-prioritization) 접근법입니다.
본 연구팀은 FUSION 파이프라인을 이용해 위 조직의 각 세포 유형마다 cell type specific TWAS를 수행했습니다. 파이프라인은 세 단계로 진행됩니다. 먼저 각 세포 유형에서 모든 세포 중 1% 이상의 세포에서 UMI가 1개 이상 검출될 만큼 발현된 유전자만 남기고, 그 유전자의 TSS ± 1 Mb 창에 품질 관리(QC)를 통과한 SNP가 존재하는지 확인해 1차 후보를 만듭니다. 이어서 이 후보 유전자마다 GCTA-GREML의 REML 알고리즘을 실행해 cis-SNP 유전력(h²)을 추정한 뒤, h² = 0이라는 영가설을 우도비 검정으로 평가합니다. 마지막으로 h² > 0.04이면서 P < 0.05인 유전자만 ‘cis-heritable gene’으로 확정해 TWAS 모델 학습과 검정에 사용합니다. 본 연구에서는 FUSION 파이프라인을 돌린 결과, 총 7,197개 유전자(세포 유형별로 449~1,078개)가 cis-heritable gene으로 선별되었습니다. 이어서 각 유전자에 대해 동일 구간의 SNP와 세포 유형별 발현치를 연결하는 예측 가중치(reference panel)를 구축했는데요. 여기서 예측 가중치(predictive weights, reference panel)란 특정 유전자의 cis 영역(TSS ± 1 Mb)에 존재하는 각 SNP가 그 유전자의 발현치를 얼마나 설명하는지를 계수 형태로 담은 벡터 w를 말합니다. 이후 연구팀은 single best eQTL(top1), best linear unbiased prediction(blup), LASSO, Elastic Net 총 네 가지의 정규화 선형 모델을 적용해 아래와 같은 형태로 유전자 발현(y)을 개별 SNP 유전자형 행렬(X)에 회귀시켰습니다.

여기서 WGE가 바로 SNP-발현 예측 가중치이며, 5-fold 교차검증으로 가장 높은 R^2를 주는 모델의 가중치가 최종 저장됩니다*. 가장 높은 R^2(결정계수, 모델이 실제 변동을 몇 % 설명했는지를 의미)을 주는 모델이 결국 예측한 발현치가 실제 발현지를 가장 잘 설명하는 모델이기 때문입니다. 이렇게 얻은 가중치가 담긴 파일과 해당 집단의 LD 행렬(V)만 있으며, GWAS 요약통계 z-score 벡터(z)와 내적해 아래와 같이 '유전적으로 예측된 발현량(GReX)-형질 연관성'을 계산할 수 있습니다. 아래와 같은 식으로 말이죠.
* 5-fold 교차 검증에서는 고유 유전력이 높은 1000개 유전자를 무작위 추출해 모델 예측력을 평가했고, 이때 성별, 연령, H.pylori 감염 여부, 10개 유전적 PC, 2개의 PEER factor를 공변량으로 포함해 잔차화(residualization)했습니다.

이는 실제 발현 데이터를 측정하지 않고도 각 세포 유형별로 유전자-질환 연관 신호를 추적하게 해주는 유전적 스캐폴드 역할을 합니다. 본 연구팀은 그렇게 최종적으로 얻은 세포 유형-특이 가중치를 위암 GWAS 요약 통계와 결합해 각 유전자의 발현-질병 연관 통계량을 계산했으며, FDR < 0.05를 충족하는 유전자를 유의 TWAS hit으로 정의했습니다. 결과적으로 13개의 위 세포 아형에 걸쳐 GC risk와 상당히 연관된 15개의 유전자를 파악했으며, 예로 parietal cells에서 MUC1 유전자의 상향 조절과 연관된 유전변이(rs4971066의 T allele)는 GC risk 감소와 연관되어 있음을 발견했습니다(참고로, MUC1의 eQTL은 모든 세포유형에 걸쳐 서로 다른 allelic effect size를 보였습니다). 각 위 세포형에 대한 co-localization signals와 TWAS 결과에 대한 쿼리 결과는 scGaTE 플랫폼에서 확인할 수 있다고 하니 참고 바랍니다.

다음으로 본 연구팀은 세포 유형 특이적인 cis-eQTL 기저의 조절 메커니즘을 탐구하기 위해, scATAC-seq 데이터(3명 샘플, 2명 공공 데이터)를 사용하여 5명 기증자로부터의 정상 위 샘플(normal gastric samples)의 eSNP 위치와 open chromatin 영역 사이의 overlap 정도를 평가했습니다. 즉, 단일세포 eQTL 분석에서 규명된 lead eSNP가 실제로 각 세포형의 열린 크로마틴 영역 안에 위치하는지를 검증한 것인데요. 먼저 본 연구팀은 원시 ATAC 리드를 정렬 및 피크 호출을 한 뒤, QC를 거쳐 총 1만 여 개의 고품질 세포를 남겼습니다. 이어 Seurat-Signac의 CCA(anchor) 방법으로 ATAC 데이터를 scRNA-seq 데이터와 결합해 19개 위장 세포 유형 라벨을 ATAC 세포에 전이하고, MACS2를 이용해 세포형별(open-chromatin) 피크 세트를 재호출했습니다. 그런 다음 각 세포형에서 eQTL의 lead eSNP와 가장 가까운 ATAC 피크까지의 거리를 계산하고, 동일한 수의 무작위 SNP(randomly sampled SNPs)를 여러 차례 부트스트랩해 얻은 무작위 거리 분포와 비교했습니다. 통계적으로는 t-검정을 적용해 eSNP-to-peak 거리가 무작위보다 짧은지를 평가했으며, 결과적으로 모든 세포형에서 eSNP가 무작위 SNP보다 열린 크로마틴에 더 근접함을 확인했습니다(Fig 6B 참조).
Figure - PMC
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
pmc.ncbi.nlm.nih.gov
그 외에도 연구팀은 일부 eSNP가 여러 세포 유형의 open chromatin 영역에 공통으로 자리하는 반면, 다른 eSNP는 특정 세포 유형의 open chromatin 영역에만 존재한다는 사실을 확인했습니다. 예로, PSCA eQTL에 위치한 eSNP인 rs2978980와 rs2294008은 다양한 세포 유형에서 검출되었으며, 검출된 세포 유형 중 대부분의 경우 동일한 염색질 피크가 겹쳐 나타났다고 하는데요(위 Fig6C 참조). 이는 해당 eSNP들이 여러 세포 유형에서 접근 가능한 공통적인 염색질 피크 안에 위치해 PSCA 발현을 범세포적으로 조절할 수 있음을 시사합니다. 반면에 APOBEC2 eQTL에 위치한 eSNP인 rs207301는 parietal cells에서만 파악되었으며, 그만큼 parietal cells 내에서만 peaks가 겹쳐서 나타났다고 합니다. 종합적으로 본 결과는 단일세포 eQTL에서 발견된 조절 변이가 실제로 특정 세포형의 활성 크로마틴 영역에 자리한다는 근거를 제시했다고 볼 수 있겠습니다.

이제 결론 내용을 정리해보겠습니다. 우선 본 연구팀은 scRNA-seq 데이터와 SNP genotyping 데이터를 활용하여 위 세포 유형 조성을 파악함과 동시에 해당 세포 유형에서의 특이적인 유전자 발현 프로파일을 확보할 수 있었습니다. 또한, eQTL 분석을 통해 위세포형별로 유전자 발현을 조절하는 cell type specific eQTL을 파악할 수 있었습니다. 이때 세포 유형 특이적인 cis-eQTL 데이터를 GWAS로 파악한 GC(Gastric Cancer) associated variants와 통합함으로써 GWAS loci에서 변이에 의해 발현이 조절되는 GC risk gene을 파악할 수 있었습니다.
특히, GC risk genes(PSCA, PTGER4, ANKRD50, ERAP1, DTX4, EML4, APOBEC2)는 epithelium에서 특이적으로 공위치화(co-localize)되어 있었고, 이는 epithelium lineage(특히 mucous neck cell)가 GC etiology에 주요 역할을 함을 암시한다고 볼 수 있으며, GC가 gastric epithelial stem cells로부터 발생한다는 기존의 모델과도 일치하는 결과로도 볼 수 있겠습니다. 참고로, 위암(GC) 위험 유전자 가운데 mucous neck cell에서 공위치화(colocalization) 신호가 확인된 ERAP1은 기관 형성(organogenesis)과 줄기세포 항상성(stem cell maintenance)을 조절하는 생물학적 경로인 Hedgehog(Hh) 신호 전달계의 핵심 조절 인자로 알려져 있습니다. 따라서 mucous neck cell에서 관찰된 ERAP1의 유전적 변이는 Hh 축을 통해 세포의 stemness 프로그램을 재구성하고, 이 과정에서 위암의 발생과 초기 진행을 촉발하거나 가속화할 잠재력이 크다고 볼 수 있겠습니다.
상피 계통을 넘어 면역 세포에서도 주목할 만한 결과가 나왔는데요. 본 연구는 WASF2 eSNP-eGene 신호가 naive B 세포에서 독점적으로 공위치화됨을 처음으로 확인했습니다. 여기서 WASF2는 T 세포에서 mTOR 활성을 억제해 세포 항상성을 유지하는 것으로 알려졌지만, naive B 세포에서의 정확한 기능은 규명되지 않은 상황입니다. 따라서 B 세포 특이적 WASF2 변이가 mTOR 경로와 종양 미세환경 상호작용에 어떤 변화를 초래하는지 추가로 밝혀낸다면, 위암 발생 메커니즘을 세포 계통 전반에서 더 정교하게 이해할 수 있을 것으로 추정됩니다.

cell-type specific TWAS 분석 결과에서는 벽세포(parietal cell)에서 관찰된 특정 유전 변이가 MUC1 발현을 높임과 더불어, 위암(GC) 위험을 낮춤을 확인했는데요. 여기서 MUC1은 상피 세포막을 덮는 막관통 점액질( transmembrane mucins)로, 병원체 부착, 산화, 화학적 손상 같은 외인성 스트레스(exogenous insults)로부터 점막을 차폐하는 1차 방어막입니다. 이러한 MUC1이 충분히 발현될 경우, 위 상피가 위산 분비 과정에서 겪는 강한 산 혹은 염증 스트레스에 대한 방어력이 강화되어 궁극적으로 위암 발생 위험을 낮출 수 있습니다. 실제로 선행 연구는 rs4072037 대립유전자가 MUC1 두 번째 엑손의 대체 스플라이싱(alternative splicing)을 조절해 위 조직의 보호 기능을 변화시킬 수 있음을 보고한 바가 있습니다. 본 연구는 이러한 스플라이싱 기반 조절이 벽세포(parietal cells)에 국한된 세포 유형 특이 현상임을 처음으로 밝혀냄으로써, MUC1-매개 보호 경로가 GC senstitivity를 조율하는 세포 수준의 분자 기전을 규명해낼 수 있었습니다.
지금까지 '단일세포 eQTL 매핑을 통한 위암 위험과 연관된 세포 유형-특이적 유전자 규명'을 주제로 한 논문 내용 정리였습니다. 다음 글에서는 최근에 공부한 '김기현의 딥러닝 부트캠프 with 파이토치' 책 정리를 해보도록 하겠습니다. 감사합니다!

참고자료
1. Bian, Lijun et al. Single-cell eQTL mapping reveals cell-type specific genes associated with the risk of gastric cancer, Cell Genomics, Volume 5, Issue 4, 100812
2. ChatGPT



