Hi, I'm a TKM.

I will discuss Chapter 2 in the book <Bioinformatics Algorithms> today.
The topic is "Which DNA Patterns Play the Role of Molecular Clocks?".


In summary, plants can regulate their transcription by employing specific factors that control gene expression through the binding of particular DNA regions known as motifs.
The motif is the specific short DNA region that is placed at the start of the genes.
The author said that these regulatory motifs were not perfectly preserved because they can vary at some positions in the real world.
And it appears at least once in each of the many different regions that are scattered throughout the genome.
It contrasts with the DnaA box, which is within a relatively short interval of the genome.

Then, let's look at algorithms for motif finding.
If you master these concepts thoroughly, you may be able to uncover hidden messages within the DNA region.

So, if you succeed in finding the evening element, you can regulate the carcadian clock.
But it's not easy when motifs have so many variants.
Therefore, we may have to consider several mismatches while finding regulatory motifs.
(k, d)-motif means a k-mer motif has mismatches below d.
Then, how do I find the (k, d)-motif?

In short, by comparing the SCORE( ) of k-mer from each string, we can select the most preserved motif matrix.
SCORE( ) is defined as the number of unpopular letters.
Thus, it is crucial to minimize this score in order to reveal regulatory motifs.
Let me introduce the matrix 'COUNT( )', 'PROFILE( )' through the code results made by ChatGPT.
# Example motifs
motifs = [
"AAGC",
"TCGA",
"GGAT",
"ACTG",
"TCAG",
"CGTA",
"ATGC",
"TACG",
"GATC",
"CATG"
]
# Calculations
print("Motifs:", motifs)
print("SCORE(Motifs):", SCORE(motifs))
print("COUNT(Motifs):", COUNT(motifs))
print("PROFILE(Motifs):", PROFILE(motifs))
print("CONSENSUS(Motifs):", CONSENSUS(motifs))
->
Motifs: ['AAGC', 'TCGA', 'GGAT', 'ACTG', 'TCAG', 'CGTA', 'ATGC', 'TACG', 'GATC', 'CATG']
SCORE(Motifs): 25
COUNT(Motifs): [Counter({'A': 3, 'T': 3, 'G': 2, 'C': 2}), Counter({'A': 4, 'C': 3, 'G': 2, 'T': 1}), Counter({'T': 4, 'G': 3, 'A': 2, 'C': 1}), Counter({'G': 4, 'C': 3, 'A': 2, 'T': 1})]
PROFILE(Motifs): [{'A': 0.3, 'C': 0.2, 'G': 0.2, 'T': 0.3}, {'A': 0.4, 'C': 0.3, 'G': 0.2, 'T': 0.1}, {'A': 0.2, 'C': 0.1, 'G': 0.3, 'T': 0.4}, {'A': 0.2, 'C': 0.3, 'G': 0.4, 'T': 0.1}]
CONSENSUS(Motifs): AATG
I made it easier to understand the result by the image below.

COUNT(Motifs) counts the occurrences of each nucleotide at each position in the motifs.
And PROFILE(Motifs) creates a frequency profile matrix based on the COUNT(Motifs).
Finally, CONSENSUS(Motifs) shows the most frequent nucleotide at each position.
Another method for finding motifs is to find a median string.
It's goal is to find a k-mer Pattern that minimizes d(Pattern, Dna) over all k-mers Pattern.
They defined d(Pattern, Dna) as the sum of the distances between Pattern and all strings in Dna.
And it includes the number of unpopular letters based on rows like this:

From now on, I will introduce the algorithm 'GREEDY MOTIF RESEARCH'.
It selects the most attractive alternative at every move along a string.
Since it multiplies the number in the profile, we may have to use pseudocount to substitute zeros.

Here is an example of a pseudocount adding 1 to each element.
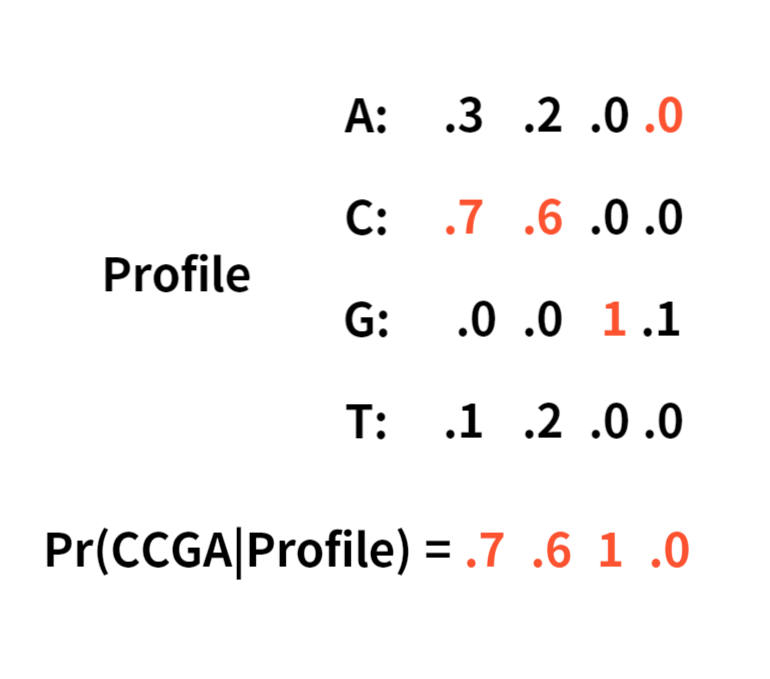

Then, let's see how the algorithm searches for the (4,1)-motif TCGC using it.
I selected 4-mer TCAC in first sequence,'ttTCACtgg'.
and computed all the probabilities of k-mer in the second sequence, ' gTAGCtaag'.
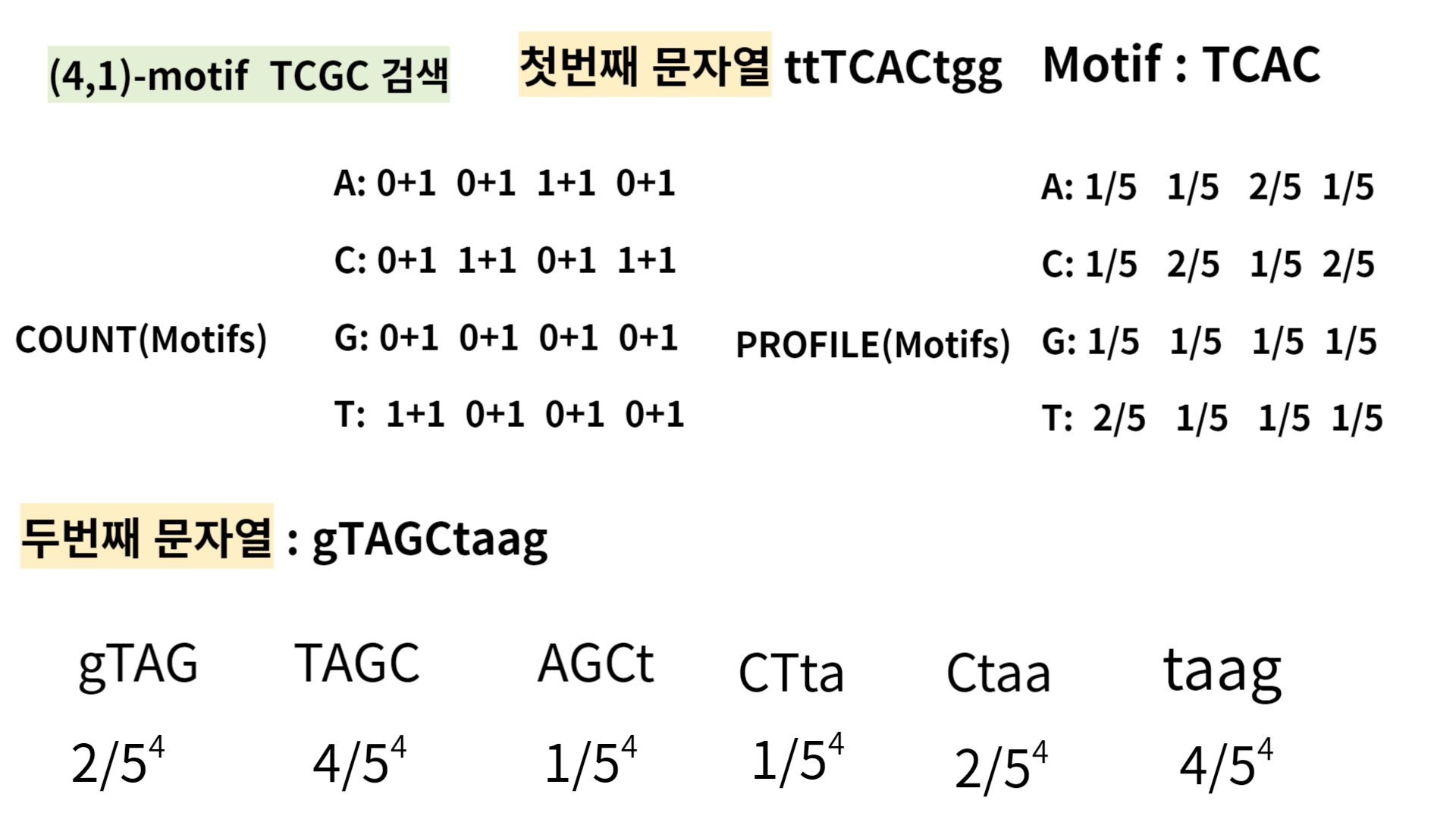
As you see, 'TAGC' and 'taag' have the highest probabilities, so pick one and compare it with the third sequence, 'gttACGCta' in the same way.
If you choose 'TAGC', you can generate a progile matrix like this:

This time, ttAC has the highest number of them.
So, by computing these probabilities for improving the profile, you will earn potential Motif effectively.
Actually, I don't know what I'm saying, so I will study it again someday.

Anyway, other algorithms such as 'RANDOMIZEDMOTIFSEARCH' or 'GIBBSSAMPLING' can find the best motif in DNA sequences.
If you want to learn about them, I recommend visiting the website below.
Bioinformatics Algorithms: Chapter 2
Learn how to find sequence motifs in DNA, hidden messages helping genes turn on and off.
www.bioinformaticsalgorithms.org
Have you heard about the disease named 'TuBerculous (TB)'?
TB is an infectious disease caused by MTB(Mycobacterium TuBerculosis).

As latent TB is related to hypoxia, biologists tried to find a transcription factor that senses the lack of oxygen and starts a genetic program.
and, in 2003, they found the dormancy survival regulator (DosR), a transcription factor that regulates many genes to adapt under hypoxic conditions.

So, it is important to discover the hidden message of DosR by trying to identify motifs within it.
That could be a key factor in targeting the infectious disease 'TB'.

과학 is 일상 : 네이버 블로그
과학기술정책가를 꿈꾸는 생물정보학도이자 과학기술계와 일반 대중을 연결하는 과학커뮤니케이터 TKM입니다! PC로 보면 좋습니다~ 공지 글로 과학꿈터뷰 전자책을 무료 공유하고 있습니다! 문
blog.naver.com
Bioinformatics OPEN STUDY | Notion
본 페이지는 비영리 목적으로 ‘생물정보학’ 관련 프로그래밍 공부 내용을 정리 및 공유하기 위해 마련해본 공간입니다. 본 페이지는 원래 비공개 페이지였지만, 일부를 공개 내용으로 바꾸는
sparkling-dibble-7ff.notion.site



