이번 글에선 전 글에서 살펴본 SNPs를 사용하는 Mendelian randomization(MR) analysis에 대해 알아보겠습니다.
What is Mendelian Randomization(MR)?
MR에 대한 간단한 설명은 아래 영상을 확인해보시면 되겠습니다.
Medelian randomization(MR)은 modifiable exposures가 health, developmental or social outcomes에 영향을 주는지 아닌지에 대한 인과관계 문제를 다루기 위해 genetic variation을 사용하는 것입니다1).
결국 Mendelian randomization(MR) analysis는 앞서 살펴본 single-nucleotide polymorphisms(SNPs)와 같은 genetic variants를 사용하여, genetic variants와 강하게 연결된 exposure과 outcome 사이의 인과관계를 조사하는 분석 방법입니다.
특히, MR의 원리는 멘델의 유전 법칙과 instrumental variable estimation method(도구 변수 측정법)을 기반으로 하기에,
confounding factor의 영향으로부터 자유롭다고 하는데, 이게 뭔 말이냐 하면 극단적으로 예를 들면 키위를 먹었을 때 A라는 질병이 호전된다는 가설을 세우고 키위를 먹어봤다고 해봅시다.
이때 A라는 질병이 호전된다고 해도, 키위를 먹기 이전에 먹었던 바나나 덕분에 A라는 질병이 호전된 것일 수도 있고, 키위를 먹은 곳의 환경이 환자의 질병이 호전되기 좋은 환경이었을 수도 있고, 그 키위가 열 등 외부 영향을 받아 변형했던 것일 수도 있습니다. 그래서 이런 요인들로부터 자유로울 수 있도록 유전자 수준에서 '노출'과 '결과' 사이의 관계를 분석하겠다는 것입니다.
비유를 계속 이어가면 말은 안되지만 그래도 극단적 비유로 특정 키위와 연관된 SNPs와 A라는 질병과 연관된 SNPs 사이에 상관관계가 있다고 분석되었다면, 이건 이전에 바나나를 먹은 영향이 들어간 것도 아니고 키위를 먹은 곳의 환경이 영향을 준 것도 아니기에 교란 요인으로부터 자유로워서 그 특정 키위(exposure)를 먹었을 때 A라는 질병(outcome)이 호전될 수 있다고 말할 수 있는 것입니다.
만약 그 상관관계가 부적 상관관계 였다면 특정 키위를 먹었을 때 A라는 질병이 더욱 악화될 수 있도 있는 것이 겠죠? 아래 영상에서 이러한 exposure과 outcome 사이의 관계를 잘 설명해주고 있습니다.
따라서 MR 분석을 활용하면 genetic variants를 통해 exposure(장내 미생물, 불면증과 같은 질병 등)의 암, 치매 등 질병(outcome)에 대한 casual effect를 분석할 수 있고,
그렇게 질병의 risk factors의 casuale role을 이해하는 것은 disease의 pahtogenesis를 파악하고, treatment strategy를 수립2)하는데 활용될 수 있습니다.
이러한 MR 분석을 정확도 높게 수행하려면 large sample size가 필요하고, genetic variant와 exposure 사이에 strong relationship이 중요하다고 하는데요,
무엇보다 risky factors(exposures)와 diseases(outcomes) 사이의 관계를 분석할 수 있는 genome-wide association studies(GWAS)의 size와 scope가 증가함으로써 MR studies를 더욱 유용하게 사용할 수 있게 되었다2)고 합니다.
허나 선행 논문2)에 따르면 confounding factor의 영향 없이 MR 분석을 통해 정확도 높게 exposure과 outcome 사이의 상관관계를 입증하기 위해선 Instrumental variable(IV) assumption이라고 부르는 세 가지 assumptions를 충족해야 된다고 합니다. 하나씩 소개해보도록 하겠습니다.
첫번째 IV assumption은?
첫번째 가정은 genetic variant는 directly하게 exposure와 연관되어 있어야 한다는 것입니다.

그래서 보통 p값이 5x10^-8 이하인 genome-wide-significant single nucleotide polymorphisms(SNPs)를 MR studies의 instruments로 활용하곤 합니다.
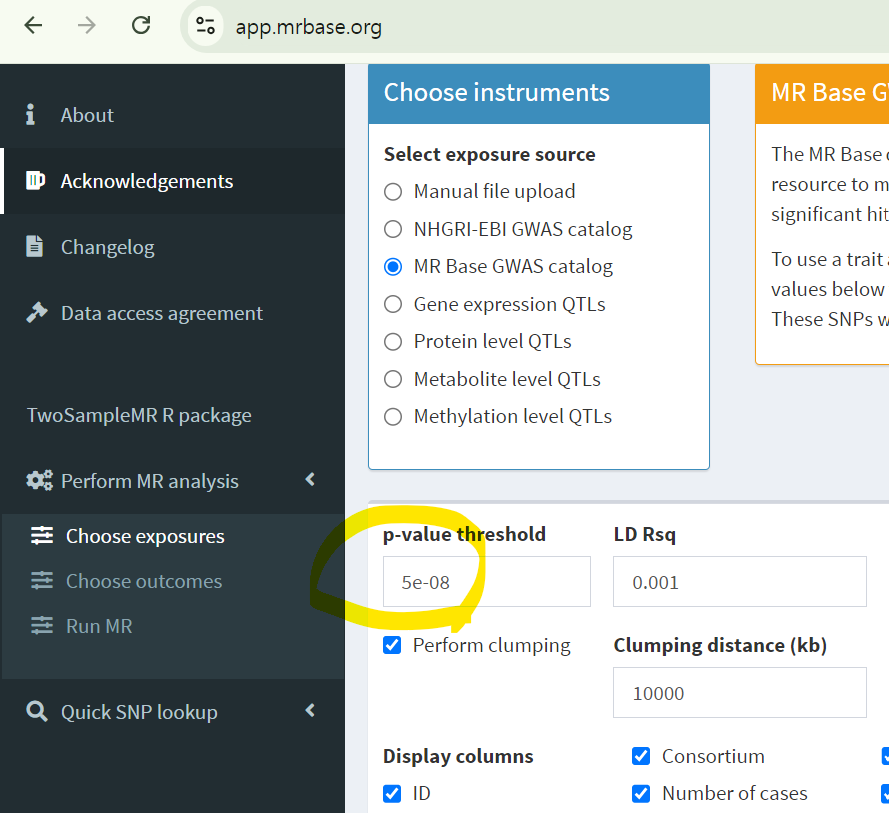
또한, 대부분 하나의 SNPs만 보는 것이 아닌 여러 SNPs를 도구 변수로 활용함으로써 MR estimates가 왜곡되는 현상을 방지한다2)고 합니다.
두번째 IV assumption은?
두번째 가정은 genetic variant가 exposure와 outcome 사이를 연결한다고 관찰된 confounding factor와 연관되어선 안된다는 것입니다.
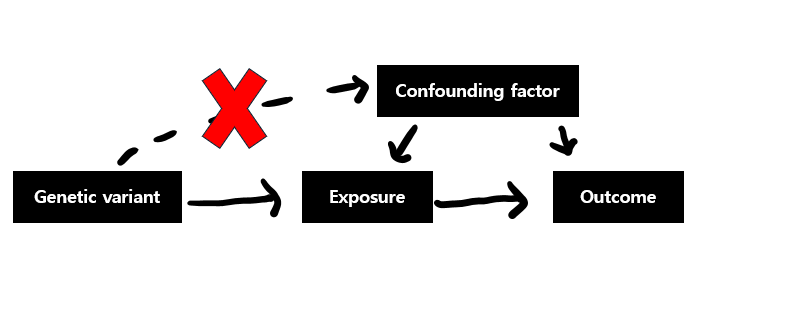
다시말해, exposure-outcome relation이 알려진 confounding factor와 variant 사이에 correlation이 있어선 안된다2)고 합니다.
세번째 IV assumption은?
세번째 가정은 genetic variant는 오직 exposure을 통해서만 outcome에 영향을 주어야 한다는 것입니다.
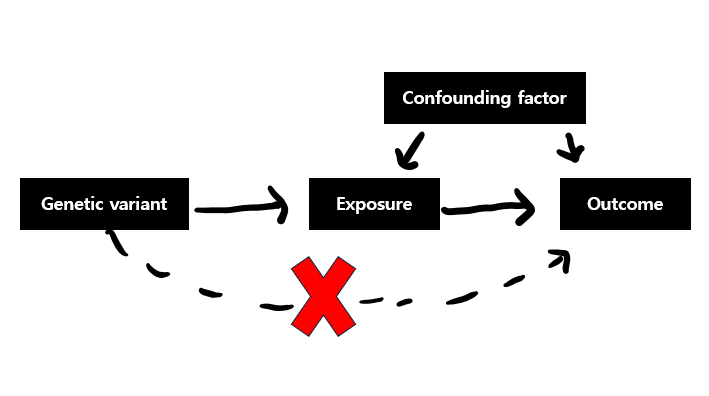
즉 pleiotropy(다면 발현 : 1개의 유전자가 그 이상의 형질 발현에 작용하는 것)가 없어야 합니다.
만약 해당 SNP가 여러 traits들과 연관이 있다면, 세번째 IV 가정은 충종되지 못할 것이기에
MR-Egger이나 weighted median test와 같은 MR design을 통해 pleiotropy의 존재 여부와 exposure의 casual effect를 확인한다2)고 합니다.
사실 선행논문2)에 따르면, 이렇게 세 가지 가정 충족한다해도 MR analysis는 여러 한계가 있는데 이에는 적절한 genetic variants의 부재, linkage disequilibrium의 존재, genetic heterogeneity, population startification(층화), confounding factors에 대한 이해 부재 등이 있다고 합니다.
여기서 linkage disequilibrium가 뭔지 궁금하실 수 있는데, 챗GPT에게 물어보니 loci A와 loci B가 chromosome에서 근접해 있다면, locus A에 있는 allele A1과 locus B에 있는 allele B1이 우연에 의한 예측우연보다 더 높은 확률로 함께 inherited되게 되는 멘델의 독립의 법칙에 위반되는 non-random association이라고 합니다(정확하지 않을 수 있습니다).

아래 영상에선 linkage disequilbrium이 어떻게 해서 발생하게 되는지 수학적 확률을 통해서도 보여주고 있습니다.
LD가 발생하게 되는 원인에 대해선 ChatGPT가 요렇게 말하는데, 비슷한 위치라서 함께 유전되기 쉬웠거나 자연 선택으로 본 combinations의 조합이 선호되게 되었을 수도 있다는 등의 얘기가 있네요.

시간관계상 대표적인 MR methods인 TwosampleMR이 무엇인지, 그리고 어떻게 측정이 이루어지는지에 대한 설명은 내일 다루어보도록 하겠습니다.
참고 자료
1) Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, Palmer T, Schooling CM, Wallace C, Zhao Q, Smith GD. Mendelian randomization. Nat Rev Methods Primers. 2:6 (2022).
2) Lee, Young Ho. Overview of Mendelian Randomization Analysis. Journal of Rheumatic Diseases. 4;27 2233-4718 (2020)




