이번 글에서는 전 글에 이어서 KOBIC '생명정보학 시작하기' 강의 3강 '미래 생물정보학자를 위한 기초 지식' 강의 내용을 정리해보겠습니다.
[20일차] KOBIC 교육강의 공부 02 :: 유전체 빅데이터
이번 글에서는 전 강의에 이어 들은 '생명정보학 시작하기'의 두번째 강의인 '유전체 빅데이터' 강의 내용을 정리해보겠습니다. 이번 강의도 개괄적인 소개를 하는 강의라 가볍고 재밌게 들을
tkmstudy.tistory.com
1. 데이터 양식
대표적인 생물정보학 데이터의 양식으로는 FASTA, FASTQ, SAM, VCF, BED가 있다고 합니다.
1-1. FASTA
먼저, 'FASTA'는 DNA 및 단백질 서열을 나타내는 가장 일반적인 형태의 데이터 양식으로, sequence 내용 전에 ID와 description을 추가할 수 있습니다 1).

위의 이미지처럼 '>'로 시작해서 sequence identifier, 그리고 sequence가 등장합니다. 그 sequence가 ' ATTAGCC..' 이런식이면 DNA 정보, ' MSKFLLBL..' 이런식이면 아미노산 정보이겠습니다.
1-2. FASTQ
FASTQ는 DNA 유전체 분석의 서열 정보 저장 양식 중 하나로, FASTA와 형식이 비슷하면서도 각 염기서열에 매칭되는 quality score를 나타낸다는 점에서 차이가 있습니다.

또한, '>'가 아닌 ' @'를 표시하고, 그 다음 염기서열 대표할 수 있는 ID, 서열, +(설명), 품질 정보가 담긴 ASVII format의 '특수 문자' 이런 식으로 이어지는데요, 여기서 읽지못한 정보는 'N'으로 등장한다고 합니다.
1-3. SAM (Sequence Alignment/Map)
SAM은 DNA 유전체 분석의 read를 참조 유전체에 매핑한 정보 저장 양식을 말합니다. 즉, FASTQ 정보를 reference genome에 매핑하여 sequence alignment한 정보 저장 양식이라고 하네요. FASTA와 FASTQ는 생물정보학 공부를 하면서 슬쩍 들어보거나 본 적이 있는데, SAM은 처음 들어봅니다.
이렇듯 매핑을 통해 SAM은 FASTQ 정보가 어떤 염색체의 정보인지, 그리고 어느 위치에 매핑되는지 알려준다고 하는데, 양식은 다음과 같습니다.

위의 read001에서 '0'은 정방향 정렬임을 의미하고, chr1은 첫번째 염색체임을 의미하고, '1007'은 참조서열에서의 시작 위치, 60은 정렬 품질 점수, 50M은 정렬의 매칭패턴(50개의 염기가 매칭), 읽기 참조 이름(*이면 없음), 읽기 시작 위치(0이면 없음), 탬플릿 길이(0이면 없음), read의 염기서열, 품질 점수(*이면 제공 X), 부가정보(ex. NM : i : 1 (서열 불일치 개수 : 1)로 메타정보들이 이어진다고 챗GPT는 말합니다.

교수님께선 "우리 몸에는 300만 개 정도 되는 변이가 존재하고, 그 변이가 차이점을 보이면서 사람들 간의 다양성을 갖게 되는데, 이 SAM을 통해서 어느 위치에서 변이가 발생하는지 볼 수 있게 된다"고 말씀하셨습니다.
1-4. VCF
VCF는 '유전 변이 정보 저장 양식'으로 변이의 header 정보, 염색체 상 존재하는 포지션, reference sequence 등을 기록하고 있습니다. 제가 봤던 SNP 데이터가 VCF 형식이었던 것 같습니다.

여기서 CHROM(변이 발견 참조 염색쳬), POS(변이 시작 위치), ID(변이 식별자), REF(참조서열의 염기), ALT(변이 서열의 대체 염기)까지는 익숙한데, QUAL(변이 호출의 품질 점수), FILTER(필터 상태), 그 외의 부가 정보들은 처음 들어봅니다.

1-5. BED
다음으로 genome annotation인 'BED'의 경우 유전체 영역을 저장하는 데이터 양식으로 유전자 위치, 유전자 이름, CHIP-seq 피크, 유전자 주변의 기능적 요소 등을 저장합니다.
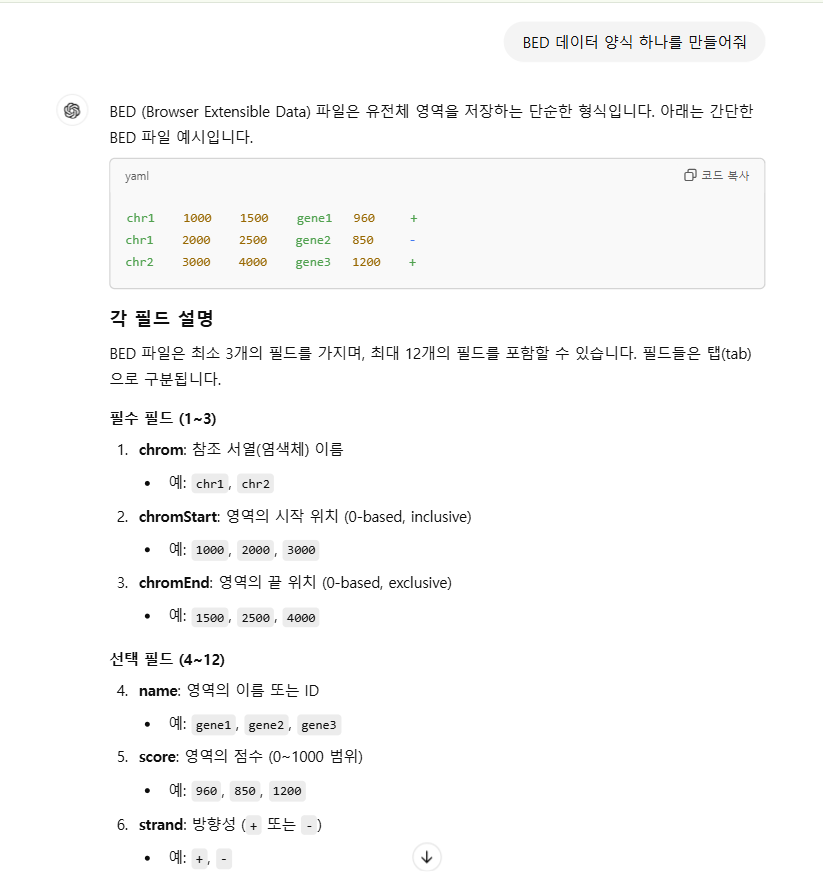
챗GPT로 BED 양식을 만들어보니 참조 염색체 이름, 유전체 영역의 위치(시작과 끝), 영역의 유전자 이름, 영역의 점수, 방향성 등을 순차적으로 표현하는 듯 합니다.
2. 프로그래밍
생물정보학 연구에 활용하는 대표적인 프로그래밍 언어로는 R, 파이썬, MAtlab, Julia, UNIX command(리눅스)가 있다고 합니다.
여기서 저는 R과 파이썬은 ADsP 자격증도 따고 프로그래밍 독학도 하고 공부를 했었는데, 다음으로 중요하다고 하는 'UNIX command(리눅스)'는 다룰 줄 몰라서, 나중에 KOBIC 교육 강의 중 리눅스 관련 강의를 들으면서 공부해봐야겠습니다. 이에 더해 아래 Bed tools를 소개하는 sandbox.bio라는 사이트에서도 UNIX 튜토리얼을 제공하는 만큼 이것도 활용해봐야겠습니다.
sandbox.bio
sandbox.bio
강의에서 교수님께서는 R의 문법 중 Tidyverse를 배워야 한다고 이야기 하셨는데, 특히 생물정보학 하는 사람들이 Bioconductor에 다가 생물정보학 할 수 있는 프로그램들을 저장해두었기에 활용하면 유용하다고 추천해주셨습니다.
Bioconductor - Home
Open source software for Bioinformatics The Bioconductor project aims to develop and share open source software for precise and repeatable analysis of biological data. We foster an inclusive and collaborative community of developers and data scientists. Ge
www.bioconductor.org
위에 사이트가 Bioconducter 사이트로, 생물학적 데이터를 분석할 수 있는 오픈소스 소프트웨어를 개발하고 공유하는 사이트인 듯하니(교육 자료도 제공하는 듯 합니다) 나중에 함 둘러봐야겠습니다.
R 문법 중 Tidyverse는 "데이터를 불러들이고, 시각화나 모델링을 위한 변형, 통계적 분석 등을 하나의 시스템 안에서 할 수 있도록 여러 함수(ex. reader, readxl, ggplot)를 통합적으로 제공해주는 패키지"라고 합니다. 과거에 R 공부할 때 학습했던 기억이 납니다.
이러한 R 프로그래밍을 공부할 수 있는 좋은 책으로 introduction to data science, advanced data science을 소개해주셨는데, 왜 좋은지는 강의를 참고하시길 바랍니다.
강의에서 교수님께서는 "생물정보학에서 중요한 것은 중요한 생물학적 질문들을 빨리 코드로 만들어서 내 눈으로 예쁘게 보는 것이며 이를 통해 성취감을 느낄 필요가 있다"고 말씀하셨는데, 생물학적 질문을 하는 방법을 익히는 좋은 R 튜토리얼인 'A Quick Start of sigminer Package'을 소개해주셨습니다.
이런 쪽 계신분들은 본인이 아는 것을 공유하는 것에 재미를 느껴셔서 그런지 몰라도 아래 링크처럼 이렇게 양질의 교육 자료를 무료로 접할 수 있습니다. 좋은 현상인 것 같습니다.
📖 Introduction | Extract, Analyze and Visualize Mutational Signatures with Sigminer
Extract, analyze and visualize mutational signatures with sigminer.
shixiangwang.github.io
물론 강의에서 교수님께서도 말씀하셨지만, 본인의 연구를 나만이 하고 나만이 아는 것이 아니라 커뮤니티에 공유하고 같은 필드에 있는 사람들이 전체적으로 높은 수준의 연구를 할 수 있도록 하는 것이 현재 생물정보학 연구에 있어 주요 가치라고 볼 수 있겠습니다. 최근에는 2024년 노벨화학상을 받은 구글 딥마인드의 작품, 알파폴드3의 오픈소스가 공개되며 화제가 되기도 했죠.
GitHub - google-deepmind/alphafold3: AlphaFold 3 inference pipeline.
AlphaFold 3 inference pipeline. Contribute to google-deepmind/alphafold3 development by creating an account on GitHub.
github.com
이외에도 생물정보학을 할 때 통계적인 것과 프로그래밍적인 것을 함께 확인해볼 수 있는 튜토리얼을 제공하는 'STHDA' 사이트를 소개해주셨습니다. 정말 디지털 사회에서는 자료가 너무 많아서 무엇이 중요한지 알기 어려운 게 탈이지, 자료가 부족해서 공부가 어려운 사회는 아닌 것 같습니다.
STHDA - Home
Statistical tools for data analysis and visualization
www.sthda.com
파이썬에서는 pandas, Numpy, Biopython를 알면 된다고 하셨는데, 관련 책으론 'Another Book on Data Science'을 소개해주셨습니다.
Biopython에선 biological computation을 할 때 활용할 수 있는 예제들을 설명해준다고 하는데요, 예로 어떻게 FASTA 정보를 parsing할지 등을 알려준다고 합니다.
Biopython · Biopython
Biopython See also our News feed. Introduction Biopython is a set of freely available tools for biological computation written in Python by an international team of developers. It is a distributed collaborative effort to develop Python libraries and applic
biopython.org
이외에도 single cell 분석을 위해 어떤 파이썬 코드로 어떻게 접근할지, 'Single-cell best practices'에서 튜토리얼을 통해 배울 수 있다고 합니다. 현재 저는 Single-cell transcriptomic analysis에 대해 알아보고자 하는 중인 만큼 관련 튜토리얼을 함 익혀봐야겠습니다.
Single-cell best practices — Single-cell best practices
www.sc-best-practices.org
생물정보학을 처음 공부하는 프로그래밍에 익숙치 않은 분들이 꼭 하는 질문은 아마 "'R'과 파이썬' 중 무엇을 배우는 것이 좋은가?"이지 않을까 싶습니다.
강의에서 교수님께선 "R과 파이썬 둘다 배우는게 좋고, 컴퓨터에 명령을 내리다가 내가 무엇을 하고 있는지 길을 잃게 되는 경우가 있는 사람이라면 R을 먼저 배우는 것이 좋다"고 말씀하셨습니다. R에서 배운 걸 파이썬에 적용할 때 개념적 접근이 더 쉽게 될 수 있기 때문이라고 하네요(변수 지정이 어렵지 않은 학생은 둘 중 무엇을 먼저 배워도 상관이 없다고 합니다).

중요한것은 본질적으로 생물정보학 데이터는 양적으로 존재하며, 이를 분석하기 위한 통계적 지식이 요구된다는 것입니다. 따라서 "다양한 기법들 중 내가 보고자하는 데이터들이 어떤 식으로 존재하고, 어떻게 데이터들로부터 의미를 찾고 결론을 찾을 수 있을지 생각해보면서 그에 맞는 통계적인 방법을 선택할 필요가 있다"라고 말씀하셨습니다.
적절한 통계적인 방법을 선택하기 위해서는 어떤 식의 통계적 접근을 하고 있는지에 대한 부분을 여러 논문을 보며 공통적인 패턴들을 볼 필요가 있다고 강조하셨습니다. 그러면서도 생물학 데이터가 어떻게 분포되어 있고, 어떤 단위로 표현되고, 발현량 분포는 시료별로 균일한지 Exploratory Data Visualization 등의 방식으로 살펴보는 것이 중요하고(내가 갖고 있는 데이터를 어떻게 생겼는지 파악하는 과정), 이를 Exploratory data analysis라고 부른다고 합니다.
더 중요한 것은 저번 2강에서도 강조했듯 현재 과학 연구는 대부분 '협업'이 필수라는 점입니다.
즉, 교수님께선 "이젠 데이터를 모으는 사람, 분석하는 사람, 결과를 해석하는 사람이 함께 일을 하게 되었다"라며 "그럼으로써 연구를 하나로 완성하면 그 연구결과를 연구분야에 있는 사람들과 함께 논의하고 평가받고 개선하는 작업들을 하게 되고, 이런 작업을 효율적으로 하기 위한 도구들이 필요해졌다"고 말씀하셨습니다. 즉, 연구를 하는 과정에서도, 연구에 대한 피드백을 받는 과정도 협업이 중요하다는 것입니다.
현재 이러한 협업을 위한 도구 중 대표적인 것이 Github라고 할 수 있으며, 협업 및 연구 재현성*을 위해 연구 분석, 개발 등에 활용한 코드를 저장하고, 버젼을 관리하는 저장소 역할을 하고 있다고 합니다.
* 연구 재현성 : 내가 어떤 결과를 관측했을 때 누구나 보고 동일하게 만들어낼 수 있는 성질 1) Method reproducibility, 2) result reproductibility
GitHub · Build and ship software on a single, collaborative platform
Join the world's most widely adopted, AI-powered developer platform where millions of developers, businesses, and the largest open source community build software that advances humanity.
github.com
정리하자면, 본 강의에서는 생물정보학 연구를 위한 데이터의 양식, 프로그래밍 언어에 대해 소개하시면서 '데이터의 분포'와 '가설 검정'을 위한 통계적인 방법을 이해하는 것이 필요하다고 하셨고, 본 프로세스에 대해 연습할 수 있는 여러가지 튜토리얼과 책들을 소개해주셨습니다. 이에 더해 생물정보학 연구에 있어 '협업'과 '연구재현성'이 필수적인 만큼 관련 도구인 'Github'를 학습하는 것도 필요하다고 하셨습니다.
정확하고 자세한 설명은 아래 INCOEDU의 KOBIC 교육센터 '생물정보학 시작하기' 3강에서 무료로 찾아보실 수 있으니, 생물정보학에 관심 있으신 분이라면 시간 되실 때 보시길 추천드립니다. 감사합니다!
차세대 생명정보 온라인 교육 | KOBIC 교육센터
KOBIC 차세대 생명정보 교육은 바이오 데이터 분석 및 활용을 위한 IT 기술(프로그래밍 언어, 리눅스)과 바이오 데이터 분석 전문기술을 제공합니다.
edu.insilicogen.com
참고자료
1) FASTA, 인코덤, URL : https://incodom.kr/FASTA
2) INCOEDU, KOBIC 교육센터, 생명정보학 시작하기

'생물정보학(바이오인포매틱스)' 카테고리의 다른 글
| [22일차] 멘델의 정원, 생물정보학 기초, Fastq QC 강의 정리 (1) | 2024.11.23 |
|---|---|
| [21일차] KOBIC 교육강의 공부 04 :: 생물정보학의 미래 기술 (24) | 2024.11.17 |
| [20일차] KOBIC 교육강의 공부 02 :: 유전체 빅데이터 (1) | 2024.11.16 |
| [19일차] 멘델의 정원 & KOBIC 교육강의 01 :: 생명정보학 시작하기 1탄 (19) | 2024.11.15 |
| [19일차] 논문 배경 지식 :: 백색 지방세포, 갈색 지방세포, 베이지색 지방세포 (15) | 2024.11.15 |



