이번 글에서는 KOBIC 교육강의 '생명정보학 시작하기'의 제4강을 정리해보고자 합니다.
[20일차] KOBIC 교육강의 공부 03 :: 미래 생물정보학자를 위한 기초 지식
이번 글에서는 전 글에 이어서 KOBIC '생명정보학 시작하기' 강의 3강 '미래 생물정보학자를 위한 기초 지식' 강의 내용을 정리해보겠습니다. [20일차] KOBIC 교육강의 공부 02 :: 유전체 빅데이터이
tkmstudy.tistory.com
4강의 주제는 '생물정보학 미래기술' 이었는데요, 인공지능, 자동화 및 컨테이너 시스템, 클라우드 컴퓨팅 순으로 소개해주셨습니다.
1. 인공지능 (AI)
인공지능(AI)은 거의 모든 분야에서 미래기술로서 적용이 되고 있지 않을까 싶습니다. 특히 현재 사회는 급속한 과학기술의 발전으로 생성할 수 있는 데이터 양이 기하급수적으로 증가하였고, 이러한 데이터 양을 처리하고 분석하기 위해서는 AI의 역할이 꼭 필요하니까요.
생물정보학에 있어 AI의 활용이 필요한 이유는 "복잡하고 다차원적 특성을 가지는 생물학적 데이터에서 유용한 패턴 및 상관관계를 찾기 위함"이라고 합니다. 이를 통해 특정 질병과 관련된 유전자 변이를 발견하거나, 단백질-단백질 사이의 상호작용 혹은 대사경로 및 유전자 조절 네트워크와 같은 복잡한 생물학적 모델링하고 분석할 수 있습니다.
또한, AI는 환자 개별 유전체 정보를 고려하여 진단, 치료, 예방 전략을 개발하는 '맞춤 의학'을 발전시키는 데 도움을 줄 것으로 기대되고 있죠. 아래 제 글에서도 생물정보학 분야의 AI 활용 가능성에 다루어보았는데, 재밌게 봐주시면 감사하겠습니다!
‘첨단바이오’의 시대, 엔비디아 등 빅테크도 뛰어드는 AI 신약 개발 경쟁이 시작되다!
AI를 활용한 신약 개발은 신약 후보 물질 발굴부터 임상시험 설계, 상용화까지 광범위한 분야에서 시간과...
blog.naver.com
이외에도 AI는 시간과 비용의 절감, 실험 계획 최적화, 데이터 수집 및 분석과정 자동화 등 생물정보학 연구 과정을 빠르고 효율적으로 이루어질 수 있도록 도움을 줄 수 있습니다. 괜히 AI가 올해 노벨과학상을 휩쓴 게 아닌 것 같습니다.
구체적인 예시로, AI 알고리즘을 통해 30억 염기쌍 중 전사 과정에 주요 역할을 하는 'DNA 속 enhancer나 promoter가 나타나는 패턴'이나 'polymerase가 DNA의 어느 지점에 붙는지에 대한 패턴'을 예측하면 기존 실험에 기인한 ATAC-seq와 같은 기술에 비해 높은 정확도와 빠른 분석 속도로 패턴을 검출할 수 있다고 합니다.

[10일차] Transcriptional Factors에 의한 gene regulation, Transcriptional Activation
Bacterial Operon에 이어 이번엔 Eukaryotes의 gene expression 조절 기전에 대해 살펴보도록 하겠습니다. [10일차] Lac Operon(structural genes, promoter, operator), 그리고 lactose와 관련된 β-galactosidase 등의 효소오늘은
tkmstudy.tistory.com
2. 자동화 및 컨테이너 시스템
'자동화 시스템'은 반복적이고 시간 소모적인 연구 과정을 컴퓨터 프로그램을 통해 자동으로 수행함으로써 생물정보학 연구자들에게 편리함과 효율성을 제공해줄 수 있습니다. 또한, 단순한 작업을 자동으로 처리함으로써 업무 품질을 향상시키고 연구재현성을 확보할 수 있다고 합니다.
생물정보학 연구에 있어 활용하는 자동화 시스템으로는 'snakemake'와 'nextflow'가 있다고 하는데 하나씩 소개해보도록 하겠습니다.
2-1. snakemake
snakemake는 "작업에서 생성되는 input, output 파일을 매개로 하는 워크플로우 관리 시스템"으로, 비순환하고 노드 간의 일방적 관계 또는 종속성을 나타내는 방향성 그래프인 유향 비순환 그래프 (Directed Acyclic Graph, DAG) 형식으로 구현됩니다. 특히, 워크플로우를 파이썬으로 구현할 수 있다 1)는 장점이 있습니다.
Snakemake
Snakemake has a powerful plugin system that allows to extend various functionalities with alternative implementations. Via stable and well-defined interfaces, plugins can evolve independently of Snakemake, and mutual update requirements are minimized. Curr
snakemake.github.io
이러한 Snakemake를 활용하면(아래 이미지 참조), "input 파일이 달라도 Task를 지정하고 흐름을 만들어주면, Input이 거기에 맞게 자동적으로 처리되어 원하는 형태의 Output(결과는 다르다 해도)이 나오게 된다"고 합니다.
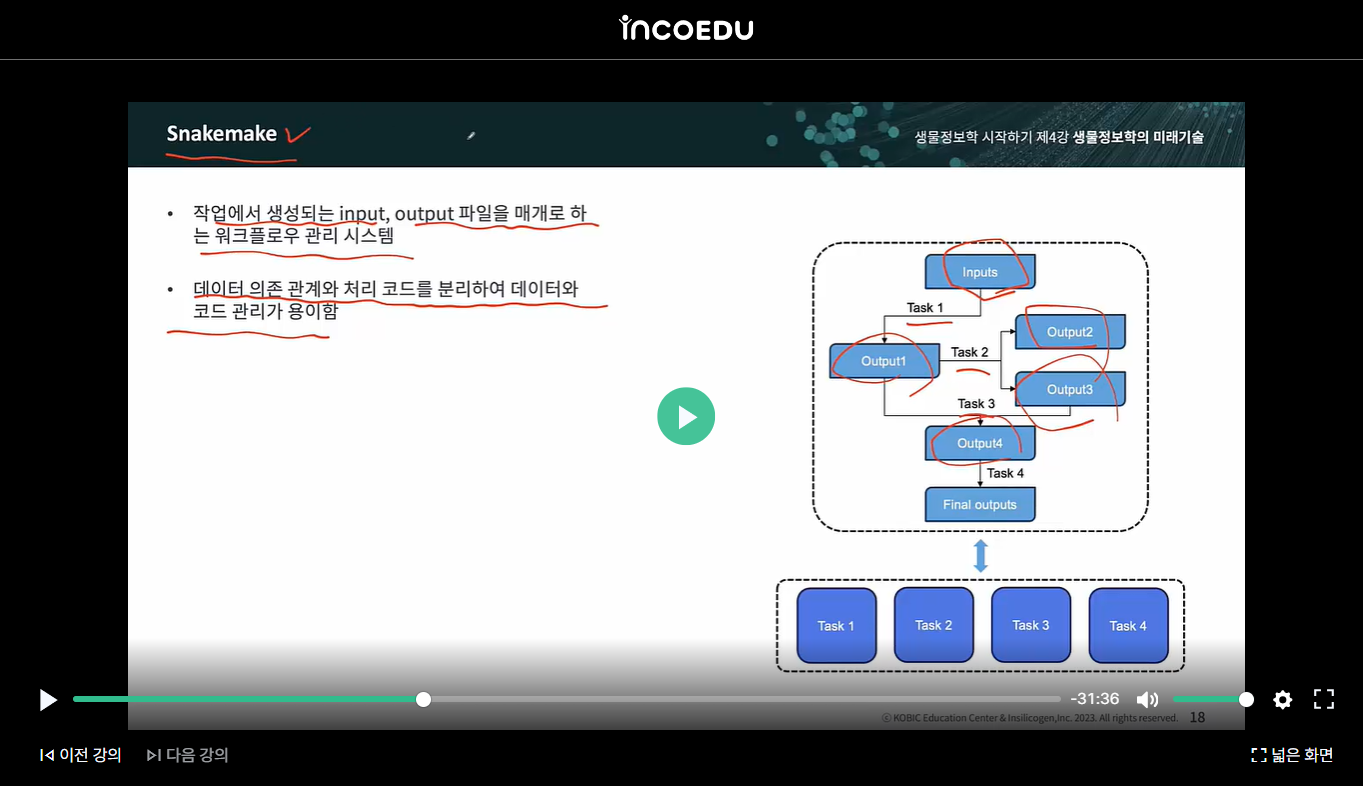
구체적으로 snakemake에서 rule을 활용해 input, output, task를 지정하면 input과 output 이름에 존재하는 variable을 매칭해서 task에 필요한 input file을 탐색하는데, 이때 input에 여러 파일을 한꺼번에 지정하기 위해 사용하는 기호인 Wildcards(와일드카드) 1)를 활용한다고 합니다.
아래는 챗GPT가 만들어준 snakemake 파일 'snakefile'의 예제입니다.
# FASTQ 파일에서 정렬(BWA) 및 변이 호출(BCFtools) 수행
# Define input and output files
SAMPLES = ["sample1", "sample2"]
# Define reference genome
REF_GENOME = "reference.fasta"
# Rule: All
rule all:
input:
expand("{sample}.vcf", sample=SAMPLES)
# Rule: Index reference genome
rule index_reference:
input:
REF_GENOME
output:
REF_GENOME + ".bwt"
shell:
"bwa index {input}"
# Rule: Align reads to reference
rule align_reads:
input:
fastq="{sample}.fastq",
index=REF_GENOME + ".bwt"
output:
bam="{sample}.bam"
shell:
"bwa mem {input.index} {input.fastq} | samtools view -Sb - > {output.bam}"
# Rule: Sort BAM file
rule sort_bam:
input:
"{sample}.bam"
output:
"{sample}.sorted.bam"
shell:
"samtools sort {input} -o {output}"
# Rule: Call variants
rule call_variants:
input:
bam="{sample}.sorted.bam",
ref=REF_GENOME
output:
"{sample}.vcf"
shell:
"bcftools mpileup -f {input.ref} {input.bam} | bcftools call -mv -Ov -o {output}"
2-2. Nextflow
또 다른 워크플로우 관리 도구 'Nextflow'는 고성능의 컴퓨팅 환경에서 작업을 분산하고 관리하는데 유용하다고 합니다. 워크플로우의 각 단계를 자동으로 분산하여 병렬로 실행해 대규모 데이터 처리작업을 효율적으로 수행할 수 있기 때문이죠.
A DSL for parallel and scalable computational pipelines | Nextflow
Nextflow enables scalable and reproducible scientific workflows using software containers. It allows the adaptation of pipelines written in the most common scripting languages.
www.nextflow.io
Nextflow도 snakemake처럼 데이터 처리 과정을 표현하고 작업 간의 종속성을 명시적으로 정의하는데, 교수님께선 Next flow의 강점은 'Docker나 Singularity와 같은 컨테이너 기술'에 있다고 하셨습니다.
컨테이너 기술은 "서로 다른 프로그래밍 환경 간의 호환성 문제를 해결하고 소프트웨어 의존성을 관리하는 기술"이라고 하는데, 생물정보학 분석(RNA-seq, ATAC-seq 등)을 하다보면 하나의 프로그램만 설치하는 경우가 없기에 호환성 문제 해결은 꼭 필요하다고 하네요!
이런 워크플로우 도구를 기반으로 하는 자동화 컨테이너 시스템을 활용하면 어떤 것들이 어떤 흐름으로 무엇을 하는지 한 눈에 볼 수 있는 것은 물론, 전처리에 들어가는 시간을 줄일 수 있다고 합니다. 결국 이제는 생물정보학 연구에 있어 "통계적 분석과 의미를 찾는 생물정보학 연구들을 더 많이 할 것 같다"고 하셨는데, 지난 강의들과 마찬 가지로 이런 워크플로우 도구에서도 역시 중요한 게 '협업'임을 강조하셨습니다.

여기서 협업은 연구실 내 연구실 간 협업을 넘어 전세계 같은 목적으로 모인 사람들의 '협업'으로, 현재 Github와 같은 플랫폼에서 수많은 국적을 가진 사람들이 다양한 워크플로우의 편의성과 파이프라인 개선하는 작업을 계속하고 있다고 하죠. 그럼으로써 "생물정보학 연구를 단순히 연구실에서만 하는 것이 아니라 외국을 가지 않아도 남대문 시장 어느 까페에서 전세계 사람들과 같이 할 수 있다"고 비유하셨습니다.
3. 클라우드 컴퓨팅
클라우드 컴퓨팅은 "서로 다른 물리적인 위치에 존재하는 컴퓨터들의 리소스를 가상화하는 기술로 통합해 제공하는 기술 3)"을 말합니다.

즉, 클라우드 컴퓨팅은 인터넷을 통해 컴퓨터 리소스나 서비스를 제공하는 기술 및 모델로, 방대한 양의 데이터 객체를 직접 다운 받지 않아도 클라우드 서비스를 기반으로 전세계 언제 어디든 해당 객체를 고유한 URL을 통해서 엑세스 할 수 있어 시간과 비용을 절약할 수 있다고 합니다.
대표적인 의생명 연구들이 클라우드를 사용하는 이유로는 다음과 같은 4가지 이유가 있다고 말씀하셨습니다.
1) 전세계 연구자들이 동시에 같은 데이터를 접근할 수 있고,
2) 내가 하는 분석을 누구나 동일하게 재현할 수 있으며(연구 재현성),
3) 데이터 공유에 들어가는 비용과 시간이 줄어들고,
4) 대규모 데이터 분석 시간 감소하기 때문입니다.
예로, gnomAD 데이터베이스(유전체 분석에서 흔히 사용되는 유전변이 빈도의 레퍼런스 데이터)의 경우 500Gb나 되는 파일을 다운로드 받아 서버에서 VCF와 병합하게 되면 총 6시간 정도가 소요된다고 하는데요.
클라우드 인프라인 AWS를 사용하면 다운로드를 할 필요가 없이 URL을 통해 전세계 누구든 언제든지 해당하는 동일한 객체에 접근할 수 있고, 단 몇 분이면 작업을 완료할 수 있다고 하죠. 따라서 많은 대형 컨소시엄, 유전체 연구 데이터가 이런 클라우드 컴퓨팅 방식을 활용한다고 합니다.
보다 자세하고 정확한 설명은 아래 INDOECU의 KOBIC 교육센터, '생명정보학 시작하기' 4강에서 확인하실 수 있습니다. 무료로 볼 수 있는 양질의 자료이니 시청하시길 추천드립니다!
차세대 생명정보 온라인 교육 | KOBIC 교육센터
KOBIC 차세대 생명정보 교육은 바이오 데이터 분석 및 활용을 위한 IT 기술(프로그래밍 언어, 리눅스)과 바이오 데이터 분석 전문기술을 제공합니다.
edu.insilicogen.com
이제 생명정보학 시작하기 강의를 총 4강의를 다 들어서 수료증이 나왔는데, 수료증이 중요한 건 아니지만 그래도 괜히 뿌듯한 것 같습니다 :)

다음으로 넘어가기전에 저번 3강에서 알게 되었던 R와 파이썬 생물정보학 관련 튜토리얼들이 굉장히 유용한 것 같아서, 하나씩 연습해보고 넘어가보려고 합니다. 또 정리할 게 있으면 찾아오겠습니다. 감사합니다!
참고자료
1) 위키덤, snakemake, 2020, URL : https://incodom.kr/Snakemake
2) INCOEDU, KOBIC 교육센터, 생명정보학 시작하기 4강
3) NAVER 지식백과, 매일경제, 클라우드컴퓨팅, URL : https://terms.naver.com/entry.naver?cid=43659&docId=17209&categoryId= 43659

'생물정보학(바이오인포매틱스)' 카테고리의 다른 글
| [22일차] 초보자를 위한 scRNA-seq data 분석법 글 추천, Edu labeling, Cre-loxp system (4) | 2024.11.23 |
|---|---|
| [22일차] 멘델의 정원, 생물정보학 기초, Fastq QC 강의 정리 (1) | 2024.11.23 |
| [20일차] KOBIC 교육강의 공부 03 :: 미래 생물정보학자를 위한 기초 지식 (21) | 2024.11.16 |
| [20일차] KOBIC 교육강의 공부 02 :: 유전체 빅데이터 (1) | 2024.11.16 |
| [19일차] 멘델의 정원 & KOBIC 교육강의 01 :: 생명정보학 시작하기 1탄 (19) | 2024.11.15 |



