안녕하세요, 이번 글에서는 멘델의 정원 유튜브 Fastq QC 관련 영상을 정리해보고자 합니다.
참고로 최근에 KOBIC 교육 영상인 '예제 데이터를 활용한 단일세포 전사체 데이터 분석'을 수강완료했습니다.

이게 보는 걸로 끝나는게 아니라 직접 해보면서 무엇인지 감을 잡아야 할 것 같은데, 본 강의에서 첨부파일로 예시를 제공해주셔서 직접 따라해볼 수 있어서 좋았습니다.
그렇지만 어느정도 따라 하다보니까 제가 윈도우 환경에서 R이랑 파이썬 언어만 사용해본지라 아직 익숙치 않은 리눅스 환경이랑 커맨드라인에 친숙해져야 할 것 같다는 느낌이 들었습니다. 사실 R이랑 파이썬도 살짝 까먹은 감이 있어서 최근에 제가 예전에 올려둔 블로그 글들 다시 보며 복습했습니다.
[개요] 데이터 사이언티스트를 위한 데이터 분석 유형 정의, 그리고 모델링 위한 5단계 프로세스
지금까지 저는 파이썬과 R을 기반으로 하는 프로그래밍 기초 지식들에 대해 학습하고 있었습니다. 하지만 ...
blog.naver.com
암튼 우선, Virtual Box로 Ubuntu 환경을 만들어둔 만큼 여기서 리눅스 관련 연습을 해보고, 커맨드라인도 관련 공부자료를 찾아서 알아봐야겠습니다. 배워야할 게 정말 많은 듯 합니다!
따라서 일단 해당 강의 정리는 직접 해보는 걸 완료한 후 나중에 해보도록 하겠습니다. 그 전에 생물정보학 연구를 하게 된다면 루틴하게 하게 된다는 'Fastq QC'를 하는 법에 대한 강의를 정리해보겠습니다.
항상 그래왔듯이 Fastq QC가 무엇인지 챗GPT에게 물어봤습니다.

즉, 앞선 글에서 정리햇듯 FASTQ 파일에는 DNA 또는 RNA 시퀀싱 데이터에 품질 점수도 들어가는 만큼 Fastq QC는 이 시퀀스 데이터를 검토하여 분석에 적합한지 판단하고(품질이 낮은지, 데이터가 오염되었는지, 시퀀싱 깊이와 리드 길이가 어떠한지), 데이터를 연구 목적에 맞게 정제해(저품질 데이터 제거, 어댑터 및 바코드 시퀀스 제거) 후속 분석의 정확도를 높여준다고 합니다.
그렇다면, Fastq QC는 NGS 분석 프로세스 중 어느 단계에 적용되는 것일까요?
영상1)에서 말하길, 일반적인 NGS 프로세스는 Library preparation, Sequencing library, Raw sequencing data, Alignment, Qauntification or variant calling, 그리고 Bioinformatics analysis으로 이어지는데 여기서 분석 전 각 단계마다 ‘Quality control’을 한다고 합니다.
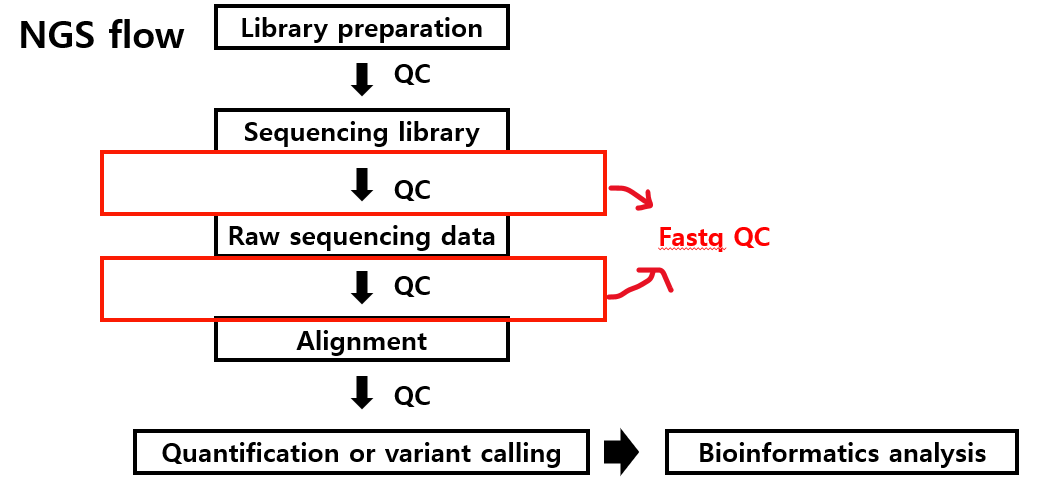
Library preparation에 대한 QC는 보통 Bioanalyzer로 진행하고, 이후 Sequencing library와 Raw sequencing data에 대한 QC는 Fastq QC로 진행한다고 하죠.
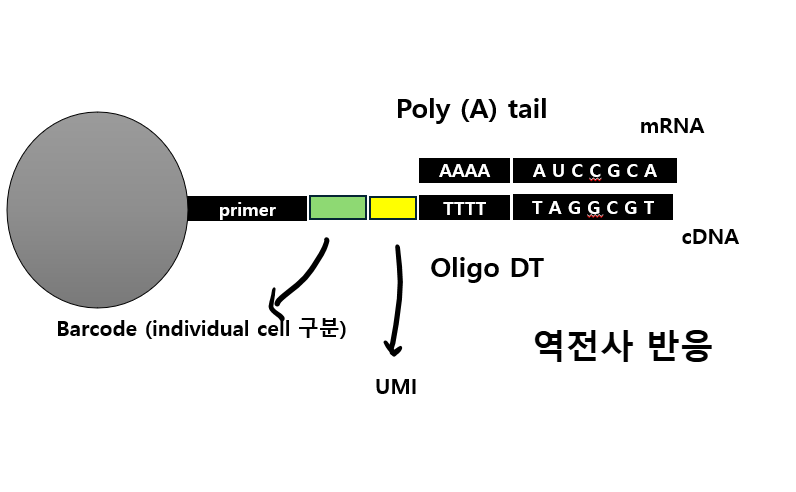
참고로, Fastq QC를 통한 품질 분석의 대상이 되는 시퀀스 리드(reads)는 DNA 혹은 RNA의 fragment가 라이브러리 후 시퀀싱이 되어 만들어지는 것으로, 시퀀싱 기기에서 한번 읽혔다는 의미에서 '리드'라고 부른다 1)고 합니다. QC 단계에선 이러한 리드의 길이와 총 수량이 적절한지 분석하여 저품질 데이터를 필터링하게 됩니다.
FastQC는 먼저 Fastq파일에 적혀있는 read에서의 GC content의 비율을 확인하는데요. 인간에겐 GC가 전체 염기서열의 약 40% 정도인데 실제 인간 데이터에서 기댓값만큼 나왔는지 확인함으로써 오류가 있는지 체크하는 것이죠.
'정빈이의 공부방' 블로그에서 본건데, GC 함량이 기대보다 너무 높으면 rRNA contamination이 있을 가능성이 높고, 기대보다 너무 낮은 경우 '역전사 반응'이 제대로 되지 않을 가능성이 높다 2)고 하는데, 그 이유에 대해선 나중에 차차 알아봐야겠습니다.


위에서 보듯 챗GPT가 말하기론, GC 함량이 너무 높으면 rRNA가 일반적으로 GC 함량이 높기에 rRNA가 너무 많다는 의미로 불필요한 rRNA가 시퀀싱에 과도하게 읽힐 가능성이 있고 (과도하게 발현되진 않았지만 중요한 시퀀스 정보를 파악하지 못하게 될 가능성 O),
GC 함량이 낮으면 샘플이 AT-rich한 특성을 보여 결합이 이중결합인 만큼 (GC는 삼중결합) 구조적 안전성이 낮아 상보적 결합이 더 쉽게 깨질 수 있고, 그럼으로써 역전사효소가 cDNA를 합성할 때 효율적으로 서열을 복제를 하지못해 '역전사 반응' 실패 가능성이 존재한다고 합니다 (일반적인 NGS 분석을 하려면 역전사 반응은 필수적이니까요).
FASTQC에서 GC비율을 확인할 수 있는 base 당 sequence content는 요렇게 표현해줍니다.
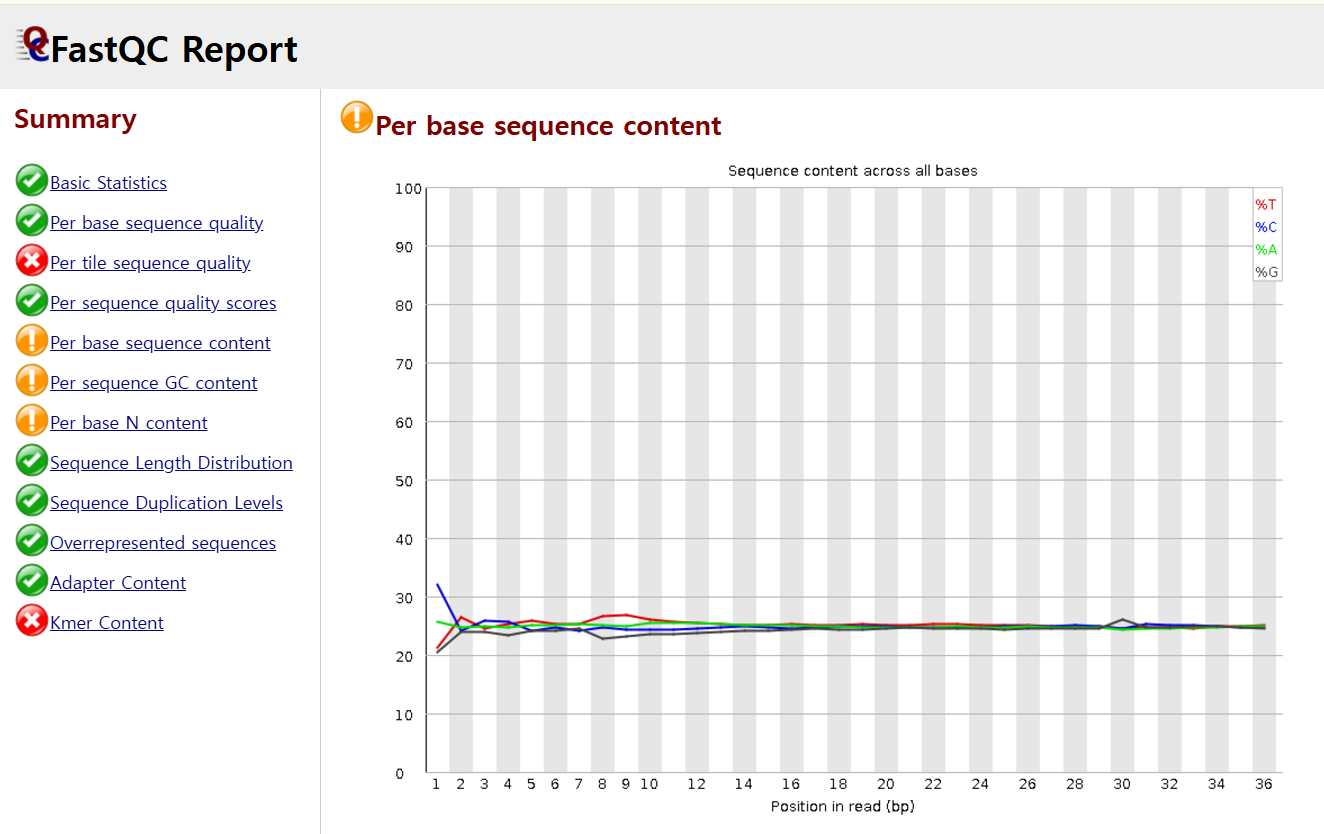
참고로, 위에는 FASTQC report인데, 왼쪽에서 보이는 각 체크 항목들에 적합한지 초록색, 노란색, 빨간색을 통해 알려줍니다.
영상에서는 오른쪽엔 A(연두)- T(빨강) 60%, C(파랑)-G(회색) 40% 이렇게 일치해서 나오기도 하지만 위 왼쪽 이미지에서 보듯 read의 앞 부분 위치에 요상한 형태가 등장하기도 한다고 하셨습니다.
그 이유는 "라이브러리를 만들 때 다양한 회사에서 만든 키트를 사용하는데, 각 키트마다 RNA ligation이 잘 되도록 하는 고유의 라이브러리 start position 즉, enzyme과 반응이 잘되도록 하는 특정 염기서열을 갖고 있을 수 있기 때문"이라고 하네요.
그만큼 공동연구를 할 때 좋은 습관은 어떤 키트를 사용했고, 그 키트의 특징은 무엇인지 생각하는 것이라 말씀하셨습니다.
이러한 특이적으로 나타나는 부분은 짜르지 않고 STAR이나 Salmon을 사용하여 alignment하면 그 안에서 클리핑이 되어 quantification 문제가 생기지 않지만, RNA-seq으로 isoform을 찾거나 exon junction의 차이를 보고자 하면 분석 전 그냥 매핑을 해서는 안되고, 파라미터 조정 등의 분석이 필요하다고 하죠.
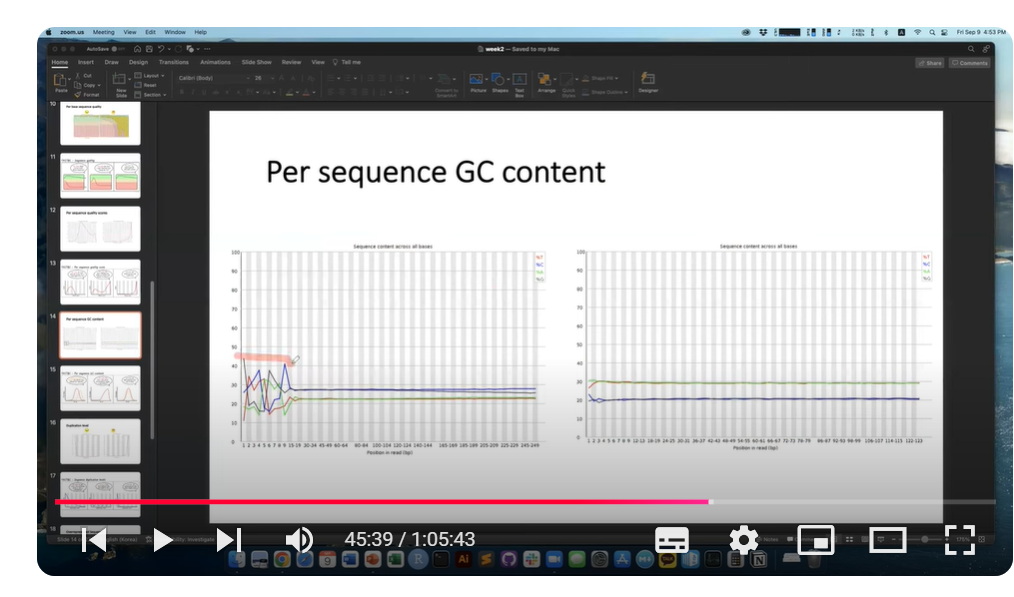
Per sequence GC content로도 시퀀스 당 GC 비율의 분포를 확인할 수 있습니다.

FASTq QC는 GC 비율만 확인하는 것이 아닌 시퀀스 리드의 길이, 수량 등 여러 체크사항들이 있죠. 아래는 Per base sequence quality인데, x축은 base pair의 위치 첫번째부터 36번째까지 나타낸 것이고, y축은 quality score로 -log10의 스케일로 나타낸 것입니다.

이에 따라 y가 10이면, 10분의 1을 의미하고, 20은 100분의 1입니다. 결국 20이면 100번을 하게 되면 1번 에러가 나타날 수 있는 확률을 갖고 있다고 볼 수 있습니다. 따라서 숫자가 클수록 에러 확률이 낮다는 것을 의미합니다. 1000분의 1, 만분의 1 이렇게 분모가 커질테니까요.
영상에서의 figure(아래 캡처 참고)를 보면, x가 커질수록 에러 확률이 상대적으로 높아지는 것을 볼 수 있습니다.

그 이유는 "일루미나의 경우, 한 cycle씩 시퀀싱을 하는데 첫번째 cycle에서 첫번째 base pair를 보고 두번째 cycle에선 두번째 base pair를 보는 방식으로 순차적으로 이루어지기에, enzyme을 이용해 화학 반응을 보는 시퀀싱이 cycle을 거듭할 수록 화학반응의 정확도와 활성도가 떨어지면서 오류확률이 높아지게 되는 것"이라고 합니다. 물론, 요즘엔 Kit가 잘 되어 있어서 Per base sequence quality가 좋은 편이라고 합니다.
결국 NGS 분석을 위해서는 분석 기기의 효소(enzyme) 관리가 중요하고, 따라서 NGS 기기에선 효소를 프레쉬하게 보존할 수 있도록 하는 냉각 시스템이 들어있다고 하죠.
다음으로 Per sequence quality score의 경우, cycle에 상관없이 전체적인 퀄리티의 분포를 보여주는데, 아래는 좋은 상황으로 read당 average quality score가 높게 나온 것(약 40)이 많다는 걸 볼 수 있습니다(x축 : score, y축 : count).
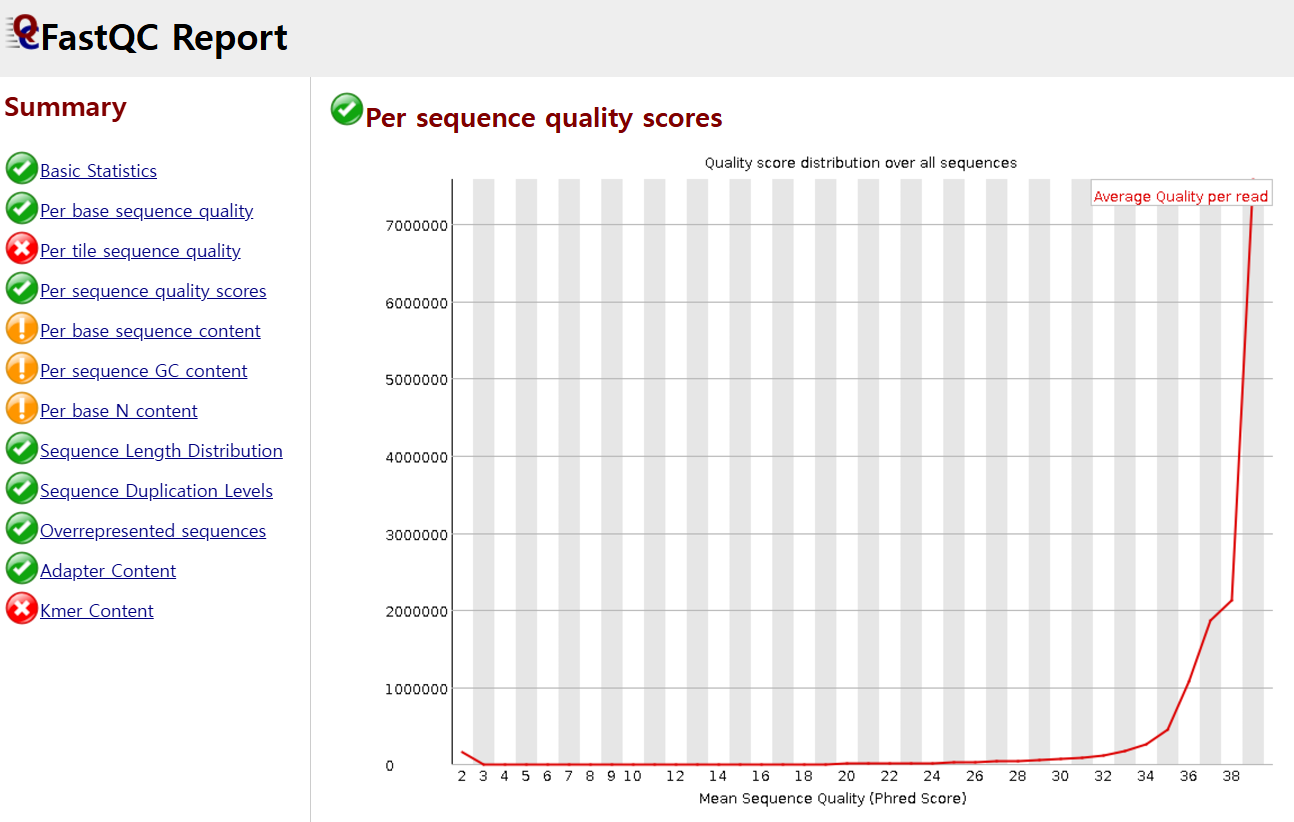
만약 퀄리티가 떨어지는 sequence read들이 등장했다면, 그것들은 Cut adapt라는 툴을 활용하여 특정 퀄리티 숫자를 기준으로 cut할 수 있다고 합니다. 이런 툴들의 라이센스를 보면 보통 MIT license인 것 같습니다.
cutadapt/doc/guide.rst at 9276a89d5df9282ff51602b042b40c3b48c98566 · marcelm/cutadapt
Cutadapt removes adapter sequences from sequencing reads - marcelm/cutadapt
github.com
이외에도 read의 중복 비율도 확인해야 하는데, sequence duplication이 발생하는 이유는 라이브러리 제작 과정에서 PCR 증폭이 이루어지기 때문입니다. 이러한 기술적 요인으로 발생한 artifact가 많으면 생물학적 차이를 파악하기 어려울 수 있으므로, duplication level이 높지 않도록 QC를 수행해야 합니다. 아래는 중복 수준이 1~2인게 대다수이게 문제가 없어 초록색 체크 표시가 떴습니다.
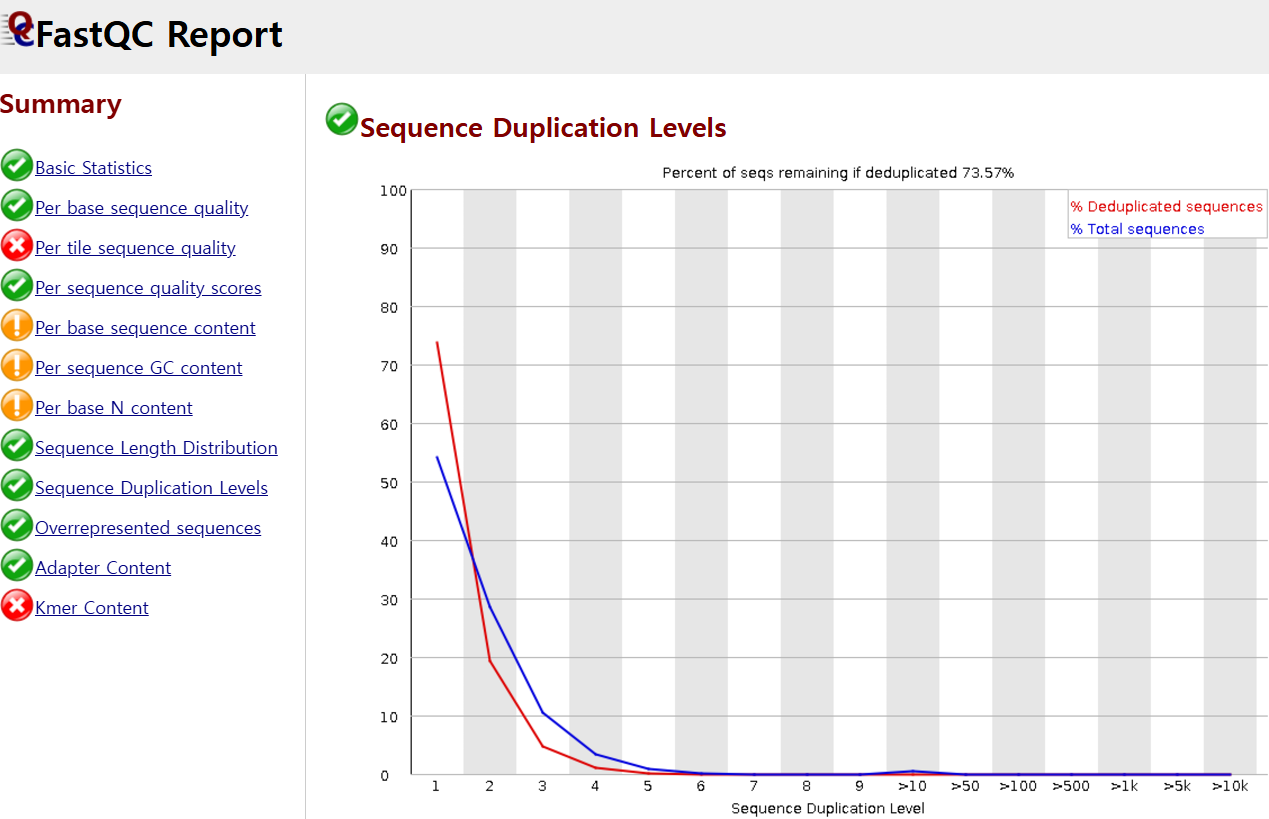
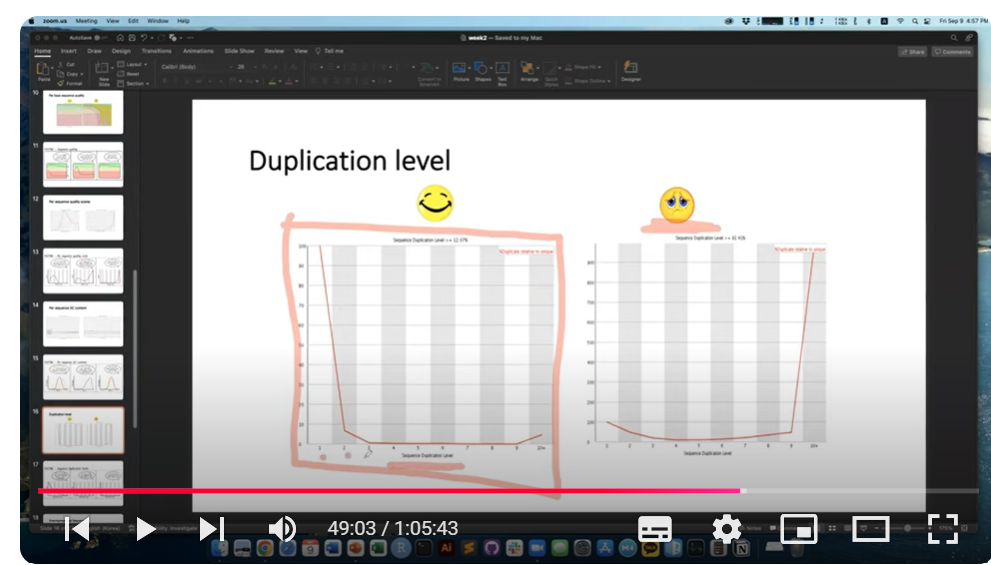
그렇지만, 아래 캡처 영상 오른쪽 이미지에서 보듯, duplication level이 10이 되는게 대다수를 차지한다면 deduplication 절차가 추가적으로 필요할 것으로 보입니다.
다음으로 overpresented sequence를 확인하는데요, 그것이 adapter일 경우 아래 캡처처럼 표현된다고 하죠. adapter가 잡힐 경우는 보통 시퀀스 input이 적을 경우인데, 실험적으로 이를 제거할 수 있는 방법이 있다고 합니다.

Non-adapter sequence도 ORS로 나타날 수 있다고 하는데 rRNA 시퀀스나 mitochondiral 시퀀스가 나올 수도 있고, 무엇인지 궁금하면 NCBI의 blast에 들어가서 species 지정 후 서치하면 확인할 수 있다고 합니다)그렇게 서치하면서 상위 몇 개의 샘플을 보며 감을 잡는 게 좋은 습관이 될 수 있다고 합니다).
Nucleotide BLAST: Search nucleotide databases using a nucleotide query
Restore default search parameters General Parameters Max target sequences ♦Max target sequences non-default value 10 50 100 250 500 1000 5000 Select the maximum number of aligned sequences to display Help Maximum number of aligned sequences to display (t
blast.ncbi.nlm.nih.gov
자세하고 정확한 설명은 글 첫부분에 링크된 영상을 확인하시면 되겠습니다. 그럼 또 다시 정리할 부분이 생기면 찾아오도록 하겠습니다. 감사합니다!
참고자료
1) 멘델의 정원, Fastq QC 하기 - 유전체 데이터를 얻고 나면 가장 먼저 하게 되는 것은?, 유튜브, 2024.04
2) 초록E, 【생물정보학】 전사체 분석 파이프라인(Transcriptomics Pipeline), 정빈이의 공부방, URL : https://nate9389.tistory.com/2050

'생물정보학(바이오인포매틱스)' 카테고리의 다른 글
| [22일차] 학부생, BRIC의 scRNA-seq data 분석법 글 따라해보기 01 :: Seurat 불러오기 & Quality Control (QC) (1) | 2024.11.23 |
|---|---|
| [22일차] 초보자를 위한 scRNA-seq data 분석법 글 추천, Edu labeling, Cre-loxp system (4) | 2024.11.23 |
| [21일차] KOBIC 교육강의 공부 04 :: 생물정보학의 미래 기술 (26) | 2024.11.17 |
| [20일차] KOBIC 교육강의 공부 03 :: 미래 생물정보학자를 위한 기초 지식 (21) | 2024.11.16 |
| [20일차] KOBIC 교육강의 공부 02 :: 유전체 빅데이터 (1) | 2024.11.16 |



