안녕하세요, 이제 기말고사와 기말과제도 끝이 나 종강을 한 만큼 대학원 준비를 이어서 해보고자 합니다.
대학원 준비생의 노트 ( 24. 12 ~ ) | Notion
'과학커뮤니케이터 TKM' TKM's STUDY BLOG
sparkling-dibble-7ff.notion.site
그런 일환으로 대표적인 single cell RNA-seq 실습 예제인 Bioconducter의 SingleCellExperiment 실습 예제를 따라해보고자 합니다.
실습에 있어서 전에 scRNA-seq 데이터 분석을 했던 Seurat과 동일하게 R package를 활용했습니다.
[25일차] BRIC의 scRNA-seq data 분석법 글 따라해보기 04 :: VlnPlot, BoxPlot, DotPlot, Cluster Annotiation
전 글에 이어서 이번에는 필터링하고 PCA 및 클러스터링한 PBMC 데이터를 가지고 또 다른 Plot들을 그려보도록 하겠습니다. 동향 | BRIC" data-og-description="1. 들어가며 R을 활" data-og-host="tkmstudy.tistory
tkmstudy.tistory.com
이번에는 아래 글을 따라해보면서 SingleCellExperiment를 진행했으며, 원 글에서는 쥐 망막 데이터인 MacoskoRetinaData를 사용했다면 저는 사람 췌장 (human pancreas) single-cell RNA-seq data인 MuraroPancreasData를 사용해서 R 환경에서 개별세포분석을 진행해보고자 합니다.
[같이실습] Single Cell RNA Sequencing #3 — Exercise (1)
Single Cell RNA Sequencing Exercise: SingleCellExperiment of Bioconductor
medium.com
데이터를 다르게 분석한 만큼 고려하지 못한 오류가 있을 수 있으니 양해부탁드립니다.
우선 분석을 위한 패키지를 깔아주었는데요, 글에서 한 방식으로 패키지를 까니까 오류가 떠서 구글에 ' SingleCellExperiment'를 검색해서 아래 사이트의 다운로드 코드를 복사해서 붙여넣었습니다.
SingleCellExperiment
Defines a S4 class for storing data from single-cell experiments. This includes specialized methods to store and retrieve spike-in information, dimensionality reduction coordinates and size factors for each cell, along with the usual metadata for genes and
bioconductor.org
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SingleCellExperiment")
참고로 제가 분석을 진행한 환경은 다음과 같습니다.
OS : Window 10
R : 4.4.2
추가적으로 BiocManager 패키지에서 사용할 수 있는 다른 패키지들도 설치해주었습니다.
BiocManager::install(c('scater', 'scran', 'uwot', 'Rtsne'))
BiocManager::install("scRNAseq")
다음으로 분석하고자 하는 데이터를 가져오고자 하는데요,
분석에 활용할 라이브러리인 'scRNAseq(SingleCellExperiemnt 설치하면서 함께 설치되었음)'에 여러 데이터가 내장되어 있어서 저는 그중 앞서 말한 사람 췌장 데이터인 MuraroPancreasData를 'sce1' 변수에 가져왔습니다.
library(scRNAseq)
sce1 <- MuraroPancreasData( )
본 데이터와 관련한 논문은 아래 링크를 참고사히면 되겠습니다.
A Single-Cell Transcriptome Atlas of the Human Pancreas - PMC
Summary To understand organ function, it is important to have an inventory of its cell types and of their corresponding marker genes. This is a particularly challenging task for human tissues like the pancreas, because reliable markers are limited. Hence,
pmc.ncbi.nlm.nih.gov
참고로 아래 글의 Reference Manual PDF를 확인하시면, scRNAseq을 통해 사용할 수 있는 다른 데이터를 알 수 있습니다.
scRNAseq
Gene-level counts for a collection of public scRNA-seq datasets, provided as SingleCellExperiment objects with cell- and gene-level metadata.
bioconductor.org
본 데이터가 어떤 형식을 띠고 있는지 확인했는데요, 뭔진 잘 모르겠지만 아마 참고한 글에서 유추해볼 때 아래 적힌 '19059 X 3072'가 유전자 19059개와 3072개의 세포를 봤다는 의미로 해석할 수 있겠습니다.

이제 Qualtiy Control을 하고자 하는데, 미토콘드리아 유전자의 비율이 높은 걸 걸러보고자 합니다.
그 이유는 세포의 자멸사(Apoptosis) 과정에서 cytoplasmic gene은 잃지만 mitochondrial gene은 그렇지 않기에 mitochondrial gene이 많다는 건 세포가 Apoptosis를 겪고 있다는 것을 의미할 수 있기 때문이죠. 이런 이유로 Low quality Cells를 필터링하는 QC 과정에서 미토콘드리아 유전자 비율이 높은 세포를 걸러야 하는 것입니다.
library(scater)
im_mito <- grepl("^MT-", rownames(sce1))
length(which(im_mito == TRUE))
결과로, '0'이 출력된 만큼 QC 과정에서 불합격한 세포는 없었습니다.
여기서 sce1의 rownames는 요런 식으로 나오는데요,

첫째 오른쪽 'A1BG_chr19'의 경우 19번째 염색체에 있는 A1BG 유전자가 사람 췌장 데이터에 등장했다고 볼 수 있겠습니다.
A1BG Gene에 대한 상세한 설명은 궁금하실지 모르겠지만 아래 글을 참고하시면 됩니다.
Gene symbol report | HUGO Gene Nomenclature Committee
All download files including the archive files are now in a publicly accessible Google Storage Bucket. Downloads page links have been updated.
www.genenames.org
미토콘드리아 gene이 없더라도 원 글에서처럼 perCellQCMetrics 함수를 이용해서 미토콘드리아 gene을 선택해 QCMetrics를 만든 후 quickPerCellQC 함수를 통해 미토콘드리아 유전자 비율이 큰 세포를 걸러보겠습니다.
qc_data <- perCellQCMetrics(sce1, subsets=list(Mito=im_mito))
filter <- quickPerCellQC(qc_data, percent_subsets="subsets_Mito_percent")
sce1 <- sce1[, !filter$discard]
다음으로 library size를 표준화하는 Normalization을 진행할텐데요, 앞서 불러온 QC를 위해 scater 패키지에 포함된 logNormCounts 함수를 이용해 표준화해보려고 합니다.
정규화 과정에서 원 글 저자가 말하길, logNormCounts를 기본 옵션으로 그대로 돌릴 경우, Log2 Transformation도 함께 진행되어 각 count의 로그값을 취하는 방식이라 라이브러리 사이즈가 완전히 같지는 않다고 하는데요. 무슨 말인진 잘 모르겠어서 챗GPT한테 물어보니 다음과 같이 답했습니다.

정리하자면, 라이브러리 사이즈를 표준화하기 위한 로그 변환 과정은 카운트가 큰 유전자의 상대적 변화는 덜 강조하고, 카운트가 작은 유전자의 상대적 변화는 더 강조할 뿐 정규화된 카운트 값의 총합과는 관계가 없어서 라이브러리 사이즈는 모든 세포에서 동일하지 않게 된다고 하는데, 맞는지는 잘 모르겠습니다. 반복되는 과정이니 나중에 차차 공부하기로 하고 우선 본 실습을 마무리 해보는 것에 집중해보겠습니다.
logNormCounts 함수로 표준화를 진행하는 코드는 다음과 같습니다.
sce1 <- logNormCounts(sce1)
count_matrix <- as.matrix(sce1@assays@data$counts[,1:10])
apply(count_matrix,2,sum)
표준화를 했으니 세포를 클러스터링하기 위해서 세포에서 발현되는 특이적인 유전자 즉, 발현량이 크거나, 분산이 큰 유전자를 찾아보는 'Feature Selection'을 해보겠습니다.
Feature Selection을 위해 먼저 앞서 설치한 scran 패키지의 modelGeneVar함수를 사용해 모든 유전자의 평균과 분산 간의 추세를 추정하고, getTOPHVGs 함수를 활용해 생물학적 변동성이 가장 높은 유전자를 선별해보겠습니다.
library(scran)
TKM <- modelGeneVar(sce1)
hvg <- getTopHVGs(TKM, prop=0.1)
뽑힌 HVG는 다음과 같이 나왔고, 개수는 총 748개였습니다.
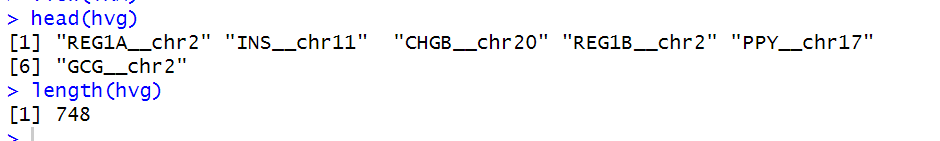
HVG(Highly Variable Genes)에 대한 설명은 아래 글을 참고하시면 되겠습니다.
[15일차] 논문 공부 :: DMEM / F12, Higly Variable Genes (HVGs), Data Normalization
이번 글에서도 저번처럼 현재 읽고 있는 논문0)을 읽으면서 중요하다고 생각되는 몇 가지 개념들에 대해 가볍게 정리하고 가도록 하겠습니다. [12일차] 논문 공부 :: Chromogenic IHC와 Immunofluroscen
tkmstudy.tistory.com
원 글에선 "유전자의 변동성은 생물학적 변동성과 기술적 편향이 합쳐진 결과이며, 이로 인해 평균 발현량이 높은 유전자일수록 분산이 높게 나타나는 '과대산포(Overdispersion)'가 나타난다 1)"라고 합니다. 과대산포가 정말 나타나는지, 모든 유전자의 평균과 분산 간의 추세를 시각화해보겠습니다.
fit.dec <- metadata(TKM)
plot(fit.dec$mean, fit.dec$var, xlab="Mean of log-expression",
ylab="Variance of log-expression")
curve(fit.dec$trend(x), col="dodgerblue", add=TRUE, lwd=2)
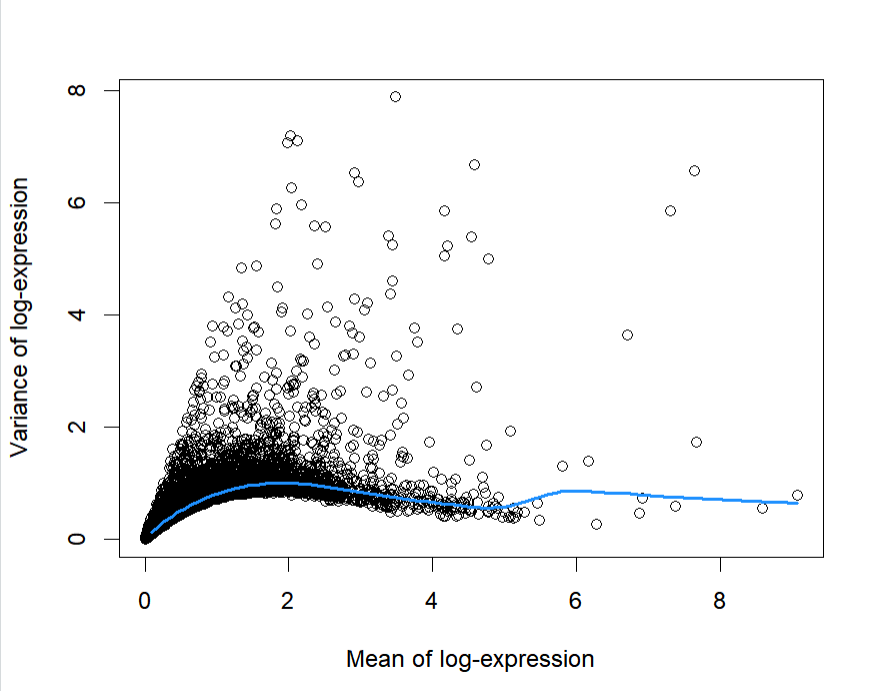
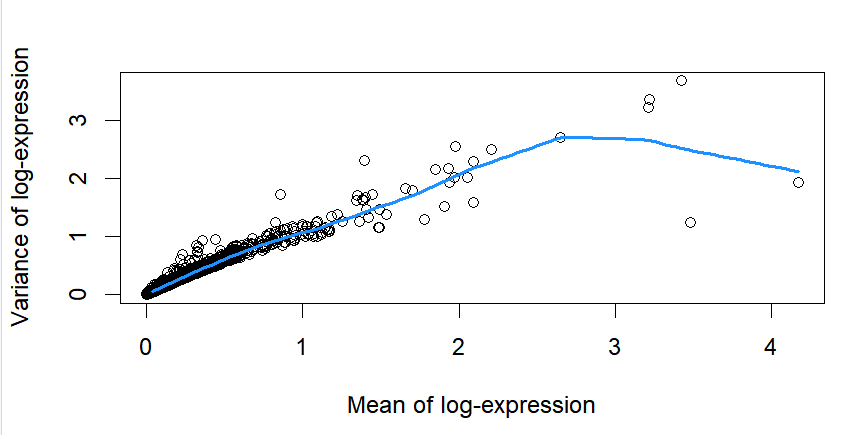
데이터가 다르다보니 원 글과 달리, 선명하게 linear한 형태가 나타나진 않은 것 같은데, 참고로 원 글과 똑같이 했을땐 다음과 같은 Figure가 등장했었습니다.
다음으로 앞서 추린 HVG를 토대로 주성분을 분석하고 차원 축소를 해보겠습니다. 이를 위해선 scater 패키지의 runPCA 함수와 runUMAP 함수를 사용해줍니다.
library(scater)
sce1 <- runPCA(sce1, ncomponents=25, subset_row=hvg)
sce1 <- runUMAP(sce1, dimred = 'PCA', external_neighbors=TRUE)
dim 함수를 이용해서 행과 열의 수를 출력해보겠습니다.
dim(sce1@int_colData$reducedDims$PCA)
dim(sce1@int_colData$reducedDims$UMAP)

결과를 보면, 2403개의 세포에 대하여 PCs(성분 개수)는 25, UMAP은 2가 출력되었음을 알 수 있습니다.
이제 최근접 이웃(Nearest Neight) 방식을 기반으로 한 buildSNNGraph 함수로 세포들을 클러스터링하고, 이를 plotUMAP 함수를 사용해 시각화해보겠습니다.
wo <- buildSNNGraph(sce1, use.dimred = 'PCA')
sce1$clusters <- factor(igraph::cluster_louvain(wo)$membership)
plotUMAP(sce1, colour_by="clusters")
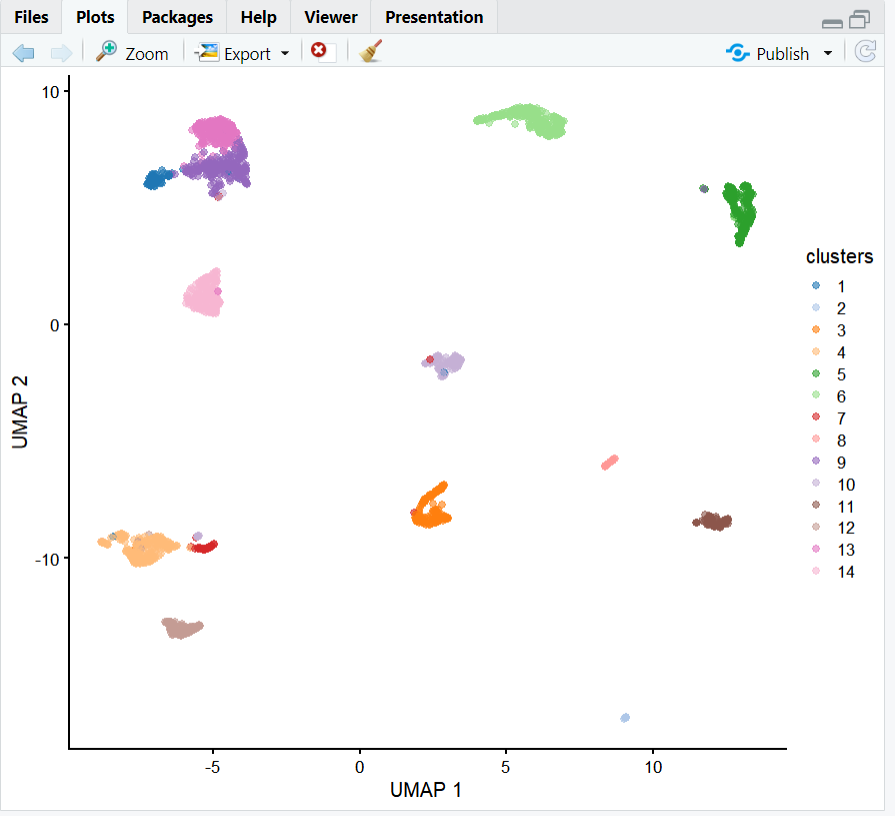
총 14개의 클러스터가 생성되었고, 서로서로 많이 떨어져있는 듯 합니다. 그럼 이것으로 Bioconducter의 SingleCellExperiment 실습을 마치도록 하겠습니다.
아직 학습해야 될 게 정말 산넘어 산입니다. 우선 쉬운 것부터 깊이 있는 이해가 부족하더라도 차근차근하다보면 single cell RNA-seq에 대해 감을 잡을 수 있지 않을까 기대해봅니다.
참고로 관련 공부에 있어 튜토리얼, 교육 자료, 데이터 포털, tool 등을 다른 글들 참고해서 노션에다가 정리해두었는데요. 이렇게 정리해두니까 보기도 좋고 사용하기도 편한 것 같습니다.
대학원 준비생의 노트 ( 24. 12 ~ ) | Notion
What to do?
sparkling-dibble-7ff.notion.site
앞으로 이곳에 정리해둔 사이트나 툴도 소개해보면서 공부해보도록 하겠습니다. 감사합니다!
참고자료
1) KH, "[같이실습] Single Cell RNA Sequencing #3 — Exercise (1)", Medium, URL : https://medium.com/biosupermarket/%EA %B0%99%EC%9D%B4%EC%8B%A4%EC%8A%B5-single-cell-rna-sequencing-3-exercise-1-cb058dace20



