이번 글에서는 KOBIC 교육강의 중 '단백질체 분석' 1편을 정리해보도록 하겠습니다. KOBIC 강의에 대한 소개는 아래 글을 참고하시면 되겠습니다.
[19일차] 멘델의 정원 & KOBIC 교육강의 01 :: 생명정보학 시작하기 1탄
이번 글에서는 우연히 알게 된 유익한 생물정보학 교육 플랫폼을 알게되어 이에 대해 소개해보고자 합니다. 오늘 어쩌다가 생물정보학 관련 유튜브 영상들을 찾아보다가 고려대 안준용 교수
tkmstudy.tistory.com
본 강의는 총 8강으로 구성되어 있으며, 황대희 교수께서 강의를 해주셨습니다. 강의는 아래 KOBIC 사이트의 '단백체 분석'에 들어가셔서 보시면 되겠습니다.
차세대 생명정보 온라인 교육 | KOBIC 교육센터
KOBIC 차세대 생명정보 교육은 바이오 데이터 분석 및 활용을 위한 IT 기술(프로그래밍 언어, 리눅스)과 바이오 데이터 분석 전문기술을 제공합니다.
edu.insilicogen.com
정리하기 앞서 단백체(Proteomes)란 "유기체(organism)에 의해 발현되는 모든 단백질의 집합 1)"이라고 정의할 수 있는데요, 여기서 단백질은 우리 몸에서 분자생물학 중심원리의 가장 끝단 즉, DNA -> RNA -> 단백질로 이어져 실제 몸의 반응을 이끌어내는 effector로서 핵심적인 기능을 하는 물질입니다.

그만큼 우리 몸을 이해하기 위해서는 단백질을 이해하는 것이 중요한데, 강의에서는 단백질 분석을 위한 Proteomics에서 주목해야 할 6가지 요인을 다음과 같이 소개하였습니다.
1) Location
2) Quantity
3) Interaction
4) Catalytic activity
5) Modification
6) Structure
정리하자면, 단백질이 1) 어디에 위치하는지, 2) 얼만큼 존재하는지, 3) 무엇(Ex. 다른 단백질)과 상호작용하는지, 4) 어떤 반응을 촉매하는지, 5) 어떤 변화를 겪었는지, 6) 어떤 구조를 갖는지 파악하는 것이 우리 몸속 단백질을 이해함에 있어 핵심 요인이라고 볼 수 있겠습니다.
첫째로 1) Location의 경우 교수님께선 "단백질의 기능은 어디에 위치하는지에 따라 달라질 수 있다"고 하셨는데, 예로 " nucleus에 주로 존재하는 transcription factor가 cytosol에서는 signaling molecule로 기능할 수도 있다"고 말씀하셨습니다.
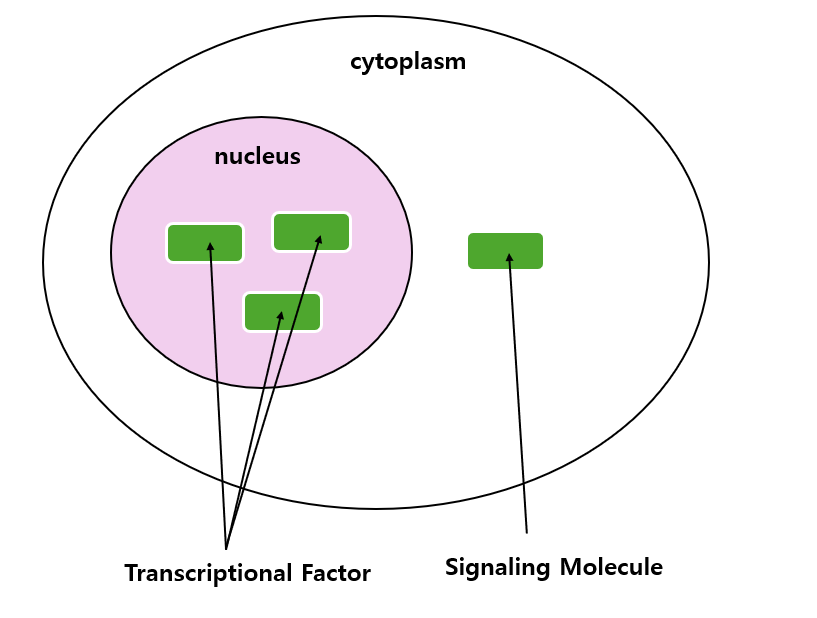
6가지 요인 중에 가장 중요한 것은 2) quantity 즉, protein의 abundance를 파악하는 것이라고 하셨습니다. 결국 생물정보학의 본질은 생물학적 데이터의 양적 특성을 파악하는 것이기에 그런게 아닐까 싶습니다.
3) interaction의 경우, 단백질은 자기 혼자 일을 할 수 없을 때 다른 단백질과 복합체를 이뤄 상호작용을 하는 만큼 본 상호작용을 이해하는 것이 단백질의 기능을 이해함에 중요할 수 있는데요. 예로, 전에 살펴봤듯 전사(trnascription) 과정에서 TF(transcriptional factor)라는 여러 단백질(ex. TFIIA ~ TFIIF)이 complex를 이루어 서로에게 영향을 주며 전사를 개시하죠.
동영상 서비스가 종료되어 해당 콘텐츠를 재생할 수 없습니다.
4) catalytic activity의 경우, 본 활성 과정을 진행하는 효소가 단백질이니까 어떤 효소가 어떤 반응을 촉매하며 세포 내 신호 전달을 활성화하는지 단백체 분석을 통해 파악한다고 합니다.
5) modification은 단백체 분석을 어렵게 하는 요인인데요, 교수님께선 "세포 내 존재하는 단백질들의 modification은 DB에 저장된 것 만 3000가지가 넘는다"라며 "하나의 단백질에서도 특정 부위엔 phospholation, 다른 부위엔 acetylation 이렇게 modification combincation이 이루어져 같은 단백질이라도 서로 다른 modification combination을 가질 수 있다"고 하셨습니다.
그만큼 단백질의 기능을 데이터를 통해 이해한다는 것은 복잡하고 험난한 길이지만, mRNA level 만으로 protein level을 추정하는 건 한계*가 있는 만큼 단백체(Proteome) 데이터를 전사체(Transcriptome) 데이터와 함께 활용하는 게 몸의 생물학적 프로세스를 이해함에 있어 정확도를 높여줌과 더불어 미쳐 생각하지 못했던 새로운 인사이트를 제공해줄 수도 있을 것입니다.
* 강의에선 교수님께서 mRNA와 protein abundance의 correlation을 구해보면 평균값이 0.3, 심지어 mRNA는 증가했는데 protein은 감소하는 경우도 보인다고 하셨습니다.

마지막 6) structure의 경우도 특정 대상이 무엇을 하는지 알려면, 그 대상이 어떤 양상으로 존재하는지 이해해야 하는 만큼 단백체 분석에서 중요한 요인으로 볼 수 있겠습니다.
한줄로 정리하자면 교수님께선 "Protein의 기능을 설명하기 위해서는 어디에 얼만큼 있으며, 어떤 다른 단백질과 interaction하고, 어떤 구조적인 change를 가지면서 catalytic activity는 어떻게 갖는지 이해하는 것이 중요하다"라고 말씀하셨습니다.
Protein의 양을 측정하기 위한 Proteomic Research를 하는 경우를 가정해봅시다. 정상 세포와 암세포의 특징을 비교해본다고 해봅시다.
본 비교를 위해서는 정상세포에는 없고 암세포에만 등장한 단백질이 있는 spot을 면도칼로 오려서 trypsin으로 digestion한 후 peptide 형태의 Fragment를 분리해 받아서 질량 분석기로 찍어보는 방법이 있다고 합니다. 그러면 Fragment의 펩티드 결합이 붕괴되면서 레더 형식의 여러 피크가 생기게 되고, 이를 이해하면 어떤 spot에서 어떤 단백질이 얼마나 있었는지 해당 단백질의 abundance를 알수 있다고 합니다.
질량분석기(mass spectrometer)에 대한 추가 설명은 아래 영상을 참고하시면 되겠습니다.
허나 질량분석기를 사용하여 단백질의 유형과 양적 특성을 파악하기 위해선 보고자 하는 단백질의 펩티드 결합이 잘 깨져야 하는데 특정 작용기의 경우 잘 안깨질 수 있다는 점에서 한계가 있다고 하네요.
그럼에도 단백질을 identify하고자 할 때 전체 단백질이 아닌 20 residue보다 적은 pepetide로부터의 sequence information이 필요하다면 질량분석이 가장 효율적이고, 10개 전후의 residue가 가장 시퀀스가 잘 된다고 알려져 있다고 합니다.
참고로 요즘엔 protein에 어떤 peptide 서열이 등장하는지 아는 상황에서 peptide level을 보고 protein level을 추정하는 bottom up 방식을 넘어 멀티오믹스를 활용한 top down (peptide level에서 추론하는 것이 아닌 protein level로 바로 확인하는 방식) 방식이 가능해졌다고 합니다.
암튼 이렇게 단백질을 identification할 때 샘플 간의 단백질 발현량 비교의 정확도를 높이기 위해 labeling을 진행할 수도 있는데요. metabolic labeling 같은 경우, 대조군과 달리 실험군에 heavy lysin을 넣어줘서 펩티드가 생성될 때 일반 lysin이 아닌 heavy lysin이 들어가도록 하여 대조군과 실험군의 단백질의 quantification과 characterization 차이를 확인할 수 있도록 하는 그런 labeling을 한다고 합니다.
대표적인 metabolic labeling인 SILAC(Stable Isotope Labeling Amino acids Cell culture)에 대한 소개 영상은 다음과 같습니다.
정리하자면 SILAC의 목적은 가벼운 아미노산이 표지된 펩티드와 무거운 아미노산이 표지된 펩티드에 무게 차이로 두개의 proteomes를 비교함에 있으며(differential enrichment, pathway analysis 사용 가능),
장점은 높은 정확도로 샘플 간 상대적 proteon level를 추정 가능하고 다양한 시스템에 적용할 수 있다는 점, 단점은 비싸고 라벨링이 불완전할 수 있다는 점이라고 하네요.
또한 iTRAQ labeling라는 것도 있는데요, 이것은 Isobaric Tag for Relative and Absolute Quantitation의 약자로 펩티드와 primay amines와 단백질을 isobaric reagents를 활용해서 라벨링하고 질량 분석기를 통해 라벨링한 reporter ion을 확인 2)하여 각 샘플의 상대적 양을 확인할 수 있다고 합니다.
이렇게 라벨을 통해 정량과 식별을 동시에 하기에 추가적인 peptide alignment할 필요가 없고, 그럼으로써 alignment 과정에서 발생하는 batch effect를 최소화할 수 있다고 하네요.
지금까지 단백체 분석 강의 1강을 정리해봤는데 강의 내용을 토대로 나름대로 요약 및 해석해서 적은 것인지라 오류가 있을 수 있습니다. 자세하고 정확한 설명은 아래 KOBIC 사이트에 들어가셔서 '단백체 분석' 강의 1강을 참고하시길 바랍니다. 감사합니다!
차세대 생명정보 온라인 교육 | KOBIC 교육센터
KOBIC 차세대 생명정보 교육은 바이오 데이터 분석 및 활용을 위한 IT 기술(프로그래밍 언어, 리눅스)과 바이오 데이터 분석 전문기술을 제공합니다.
edu.insilicogen.com
- 참고자료
1) UniProt, What are proteomes?, URL : https://www.uniprot.org/help/proteome
2) Cretive Proteomics, iTRAQ-based Proteomics Analysis, URL : https://www.creative-proteomics.com/services/itraq-based-proteomics-analysis.htm




