전 글에 이어서 이번에는 필터링하고 PCA 및 클러스터링한 PBMC 데이터를 가지고 또 다른 Plot들을 그려보도록 하겠습니다.
[24일차] BRIC의 scRNA-seq data 분석법 글 따라해보기 03 :: R 환경 Seurat의 DimPlot, FeaturePlot
이번 글에선 저번 글에 이어 BRIC scRNA-seq data 분석법 괸련 글을 토대로 KOBIC 교육영상 예제 데이터를 분석해보는 연습을 해보겠습니다. Bio뉴스 > 동향 | BRIC" data-og-description="1. 들어가며 R을 활
tkmstudy.tistory.com
앞서 말했듯 제대로된 학습을 하시고자 하는 분은 제 글을 보기보단 아래 글을 참고하시길 추천드립니다.
[당신의 논문 동료] scRNA-seq data 분석법 – Plot 4 종류부터 DEG까지 | 뉴스 > Bio뉴스 > 동향 | BRIC
1. 들어가며 R을 활용하면서 가장 많이 나오는 에러에 대해 짚고 넘어가고 합니다. 1.1. R은 대소문자를 매우 엄격히 구분...
www.ibric.org
우선, 전에 DimPlot, FeaturePlot에 이어 이번엔 scRNA-seq에서 자주 활용되는 vilolin plot을 PBMC 데이터를 토대로 그려보도록 하겠습니다.
violin plot을 그리기 위해 사용하는 함수는 VlnPlot( )이며, 전에 Featureplot에서 살펴봤던 유전자 'MS4A1'에 대한 violin plot을 그려보겠습니다.
library(Seurat) #전에 깔았던 Seurat 패키지를 로드합니다
VlnPlot(pbmc_tutorial1, "MS4A1")
* 여기서 pbmc_tutorial1이 무엇인지는 아래 글 마지막 부분을 참고하시면 되겠습니다(간단히 말하자면, 이전에 필터링하고 분석할 수 있도록 조정한 데이터를 담은 변수입니다).
[23일차] BRIC의 scRNA-seq data 분석법 글 따라해보기 02 :: Normalization부터 UMAP까지
이번 글에서는 전 글에 이어 BRIC의 scRNA-seq data 분석법 글을 따라해보도록 하겠습니다. [22일차] 학부생, BRIC의 scRNA-seq data 분석법 글 따라해보기 01 :: Seurat 불러오기 & Quality Control (QC)이번 글에
tkmstudy.tistory.com
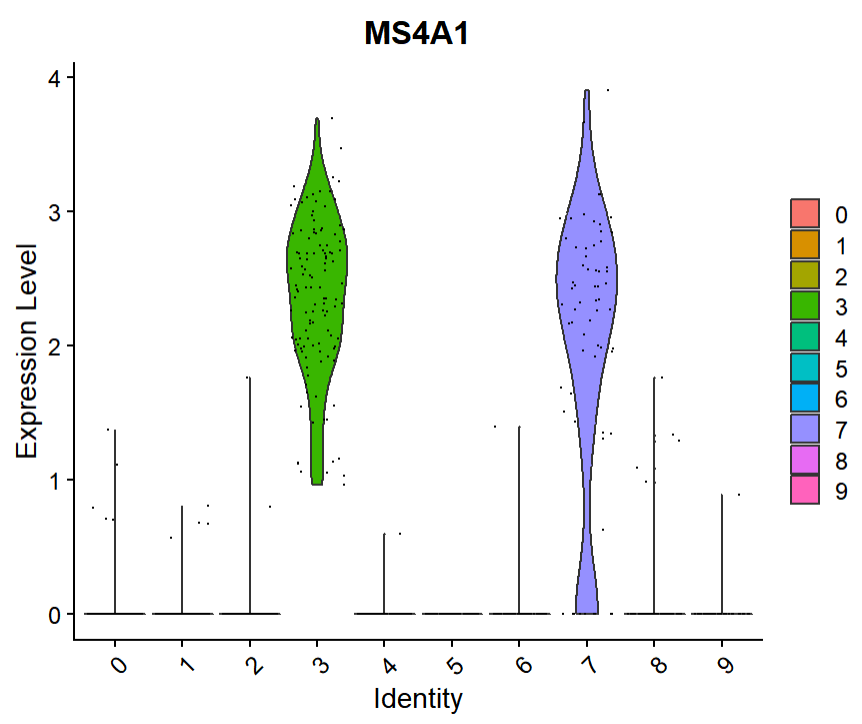
보니까 cluster 3과 7에서 M34A1이 높게 발현된 듯 합니다. Feature Plot에서도 동일한 결과를 확인할 수 있었는데, 기억 삼아 다시 불러왔습니다.
이번엔 VlnPlot( )에 보이는 점을 없애보려고 하는데요, 그러려면 점의 크기를 옵션 지정(pt.size)으로 0으로 만들면 된다고 합니다.
VlnPlot(pbmc_tutorial1, "MS4A1", pt.size = 0)

다음으로 글쓴이께서 BoxPlot을 그려보셨길래, 저도 따라서 그려봤습니다. 이를 위해서는 library( )로 불러와야 하고, 이후 geom_boxplot( )를 더해줍니다.
library(ggplot2)
VlnPlot(pbmc_tutorial1, "MS4A1", pt.size = 0) + geom_boxplot()

위에 등장한 점들은 outlier일 수 있기에 outlier.size 옵션을 통해 outlier를 없애봅시다.
VlnPlot(pbmc_tutorial1, "MS4A1", pt.size = 0) + geom_boxplot(outlier.size = 0)

다음으로 많은 유전자들의 발현량을 한꺼번에 평가하는데 활용할 수 있는 'DotPlot'을 활용해보고자 하는데요, 글쓴이가 제시한 "LYZ", "CCL5", "IL32", "FCGR3A", "PF4" 유전자가 어떤 유전자에 얼마만큼 존재하는지 확인해보도록 하겠습니다.
DotPlot(pbmc_tutorial1, features = c("LYZ", "CCL5", "IL32", "FCGR3A", "PF4")) + RotatedAxis()
여기서 '+ RotatedAxis()'한 이유는 아래 이미지에서 보듯 x축 레이블이 세로로 표시되도록 하여 레이블들이 서로 겹치지 않게 하기 위함이라고 하네요 (챗GPT 참조).

여기서 Average Expression이 진할수록 해당 유전자가 많이 발현했다고 볼 수 있고, Pecentage Expressed가 높을수록 각 클러스터에 속한 세포들 사이에서 높은 비율로 나타났다고 볼 수 있습니다. 쭉 보니 9번 클러스터에서 FCGR3A 유전자가 높은 비율로 많이 발현된 듯 합니다.
이번엔 색깔을 파란색에서 빨간색으로 바꿔보도록 하겠습니다.

x축과 y축의 전환은 해당 코드에 'coord_flip()'을 더해주면 가능합니다.
DotPlot(pbmc_tutorial1, features = c("LYZ", "CCL5", "IL32", "FCGR3A", "PF4"), cols = c("lightgrey", "red")) + coord_flip()

이제 'Cluster Annotation'을 해보려고 하는데요, 말 그대로 그 클러스터가 어떤 세포일지 생물학적 의미를 파악하고 추론하는 단계라고 볼 수 있습니다.
먼저 다시 DimPlot()을 활용해서 UMAP을 표현하고 클러스터 간 유사성을 파악해보도록 하겠습니다 (Label = T로 라벨도 표현해줍니다).
DimPlot(pbmc_tutorial1, label = T)

보니까 3번, 7번이 유사해보이고 2번 0번, 6번, 8번이 유사해보이고, 5번 1번 4번이 유사해보입니다. 참고로 UMAP에서는 가까울수록 HVGs(변동성이 높은 유전자)의 패턴이 유사하다고 볼 수 있습니다.
그리고 이젠 3,7 / 2, 0, 6, 8 / 5, 1, 4 사이의 차이를 파악하여 유사한 클러스터들의 공통된 특성을 파악해봐야 한다고 합니다.
참고로 PBMC를 대상으로 하니 (거의) 모든 세포가 면역세포일 것이고, 이는 T세포, B세포, 마크로파지, 호산구, DC, Platelet 등으로 나뉠 것이라고 하는데, 그만큼 각 celltype의 표준이되는 마커를 먼저 살펴보면 클러스터 특성을 추론하기 용이하다고 합니다.
celltype marker 유전자는 정답이 없다고 하지만, 그래도 연습이니 글쓴이가 제시한 마커를 기준으로 클러스터 분류를 해보고자 합니다.

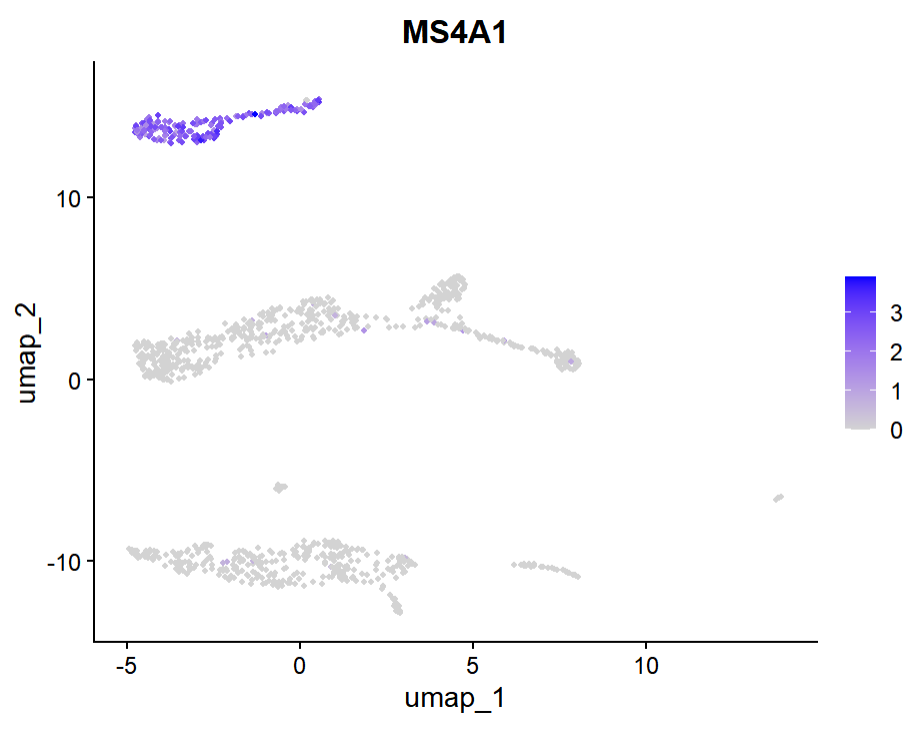
그럼 이제 클러스터들이 각 유전자를 얼마만큼 발현하고 있는지, FeaturePlot( )을 통해 확인해보겠습니다.
FeaturePlot(pbmc_tutorial1, c("CD3E", "MS4A1","LYZ", "EMR1", "CD14", "GZMA", "TREM1", "FLT3", "PPBP"))

보니까 글의 예시 데이터와 다른지라 여기엔 EMR1은 없는 듯합니다. 암튼 3,7은 B cell, 2, 0, 6, 8은 T cell, 5, 1, 4는 Monocyte라고 추론해볼 수 있겠습니다. 물론 아닐 수도 있습니다.
다음으로 이젠 클러스터의 특성을 보다 정밀하게 파악할 수 있도록 AverageExpression( ) 함수를 통해 '클러스터마다 유전자 평균 발현량'을 구해보고, 그것을 csv 엑셀 파일로 만들어보겠습니다.
Ave <- AverageExpression(pbmc_tutorial1)
write.csv(Ave, "Cluster_average.csv")
그러면 이제 요런 아래와 같은 “Cluster_average.csv”라는 파일이 설정한 디렉토리에서 만들어졌을 것입니다.

다음으로 FindAllMarkers( )라는 함수로 각 클러스터의 마커 유전자를 조사한 후 엑셀파일을 생성해보도록 하겠습니다.
여기서 FinDAllMarkers를 사용하려면 BioManager의 limma패키지를 install을 하면 좋다고 합니다. 좋다고 하니 바로 깔아보겠습니다.
install.packages('BiocManager')
BiocManager::install('limma')
Marker <- FindAllMarkers(pbmc_tutorial)
write.csv(Marker, "Cluster_marker.csv")
그러면 이제 "Cluste_marker.csv"라는 파일이 만들어졌을 것입니다. 여기에서 cluster annotation을 하기 위해서 필터를 지정하고, avg_log2FC를 내림차순으로 정리한 후 cluster 3에서 높게 발현하는 유전자를 찾아봅시다.
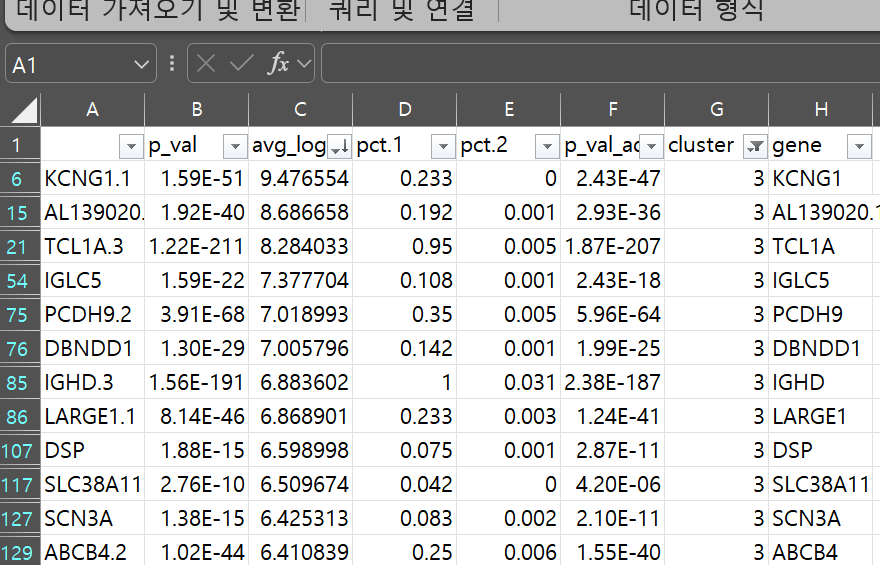
참고로, avg_log2FC는 평균 발현량 차이를 log2로 계산한 것으로 높을수록 높게 발현된 것으로 볼 수 있고, pct.1은 cluster 3 세포 중 특정 유전자를 발현하는 세포의 퍼센트, pct2.는 cluster 3을 제외한 나머지 세포 중 특정 유전자를 발현하는 세포 퍼센트를 의미합니다.
이제 scRNA-seq을 기반으로 만든 데이터베이스인 PanglaoDB를 활용해서 cluster 3에서 높게 발현한 유전자들을 검색하면 어떤 세포일 가능성이 높은지 확인해보겠습니다.
PanglaoDB - A Single Cell Sequencing Resource For Gene Expression Data
PanglaoDB is a database for the scientific community interested in exploration of single cell RNA sequencing experiments from mouse and human. We collect and integrate data from multiple studies and present them through a unified framework. Adapted from th
panglaodb.se
참고로, cluster 3에서 높게 발현한 7개 유전자는 KCNG1, AL139020, TCL1A, IGLC5, PCDH9, DBNDD1, IGHD 이렇게 나왔습니다.
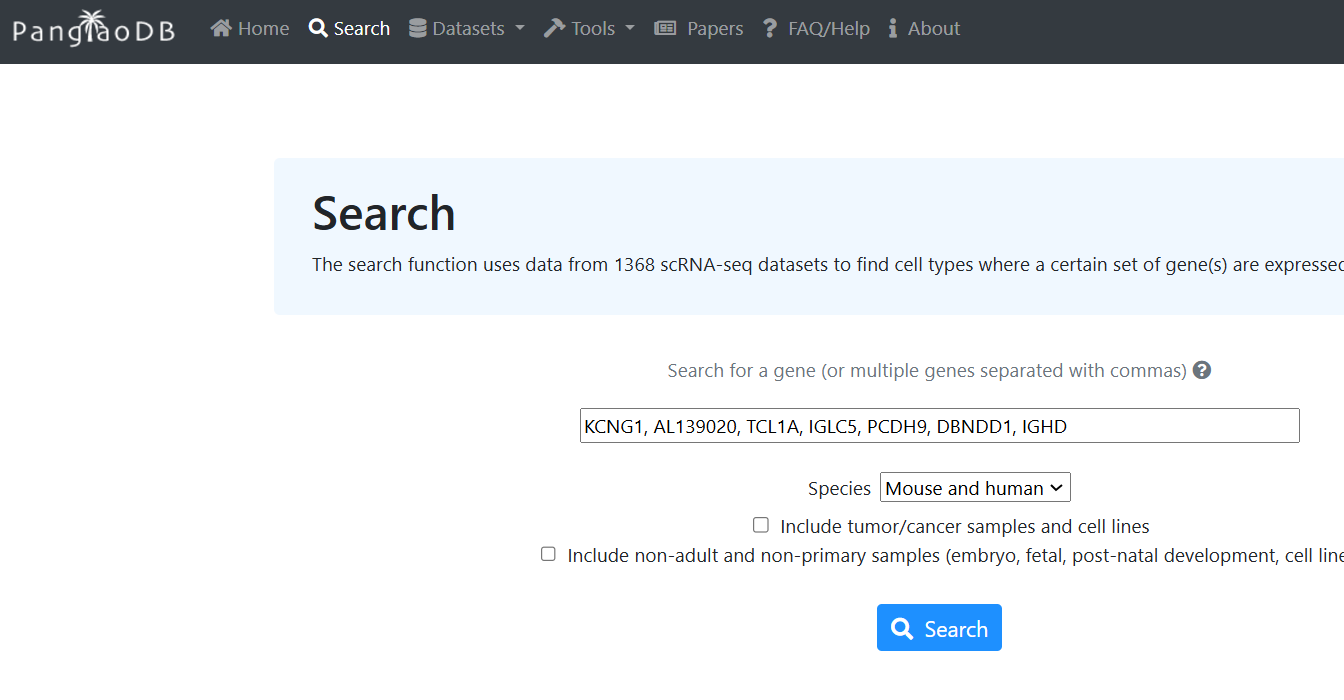
서치버튼을 누르니까 3번 cluster를 Oligodendrocyte라고 예측합니다. FeaturePlot으로 예측했던 B cell은 세번째 후보로 예측하고 있습니다.

그렇지만, 이러한 annotation은 정확하지 않을 수 있기에 여러 문헌과 데이터셋을 종합적으로 검토하는 것이 좋으며, 글쓴이께서는 cluster annotation만 1주일 가량 소요되었고 전문가분과 함께 고심해서 이름을 붙였다고 합니다.
여기까지가 BRIC 글에서 알려준 부분들을 연습한 내용이었는데요, 그 외의 배워야 할 부분들이 많은 만큼 향후 차차 공부해봐야겠습니다.
우선 이제 대학 학부생활 마지막 시험인 기말고사 준비를 할 예정이라 다시 기말 끝나고 대학원 준비 학습 및 기초체력 가꾸기를 해보도록 하겠습니다. 감사합니다!
참고자료
1) INCOEDU, KOBIC 교육센터, 예제 데이터를 활용한 단일세포 전사체 데이터 분석
2) 쿼카, [당신의 논문 동료] scRNA-seq data 분석법 – Plot 4 종류부터 DEG까지, BRIC



